Lizozomul
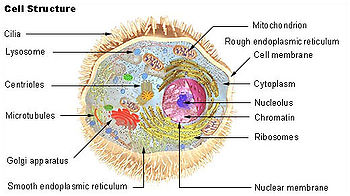
Lizozomul (din limba greacă lysis , dissolution și soma , body) este o veziculă (organet) prezentă în numeroase copii în celulele eucariote și reprezintă sistemul digestiv al celulei deoarece este responsabil pentru degradarea și digestia (distrugerea) molecule și macromolecule ingerate de celulă prin endocitoză precum și macromolecule endogene. Lizozomii se ocupă de cifra de afaceri a celorlalte organite ale celulei în sine. Prin același proces, celulele albe din sânge sunt capabile să „scape” de microorganisme patogene sau celule moarte fagocitate anterior.
Lizozomii au fost descoperite de către belgian cytologist Christian de Duve în 1949 .
Mecanism

Degradarea are loc prin intermediul enzimelor hidrolitice (numite „ hidrolaze acide ” pentru aceasta) capabile să degradeze proteinele , lipidele și carbohidrații din constituenții lor elementari și apoi, atunci când este posibil, sunt refolosite în alt mod sau expulzate.
Aceste enzime sunt activate la pH scăzut (4,8) și acest lucru este important deoarece reduce pericolul distrugerii celulei gazdă dacă există eliberarea accidentală a acestor enzime în citoplasmă (care are pH 7). Pentru o protecție suplimentară, membrana lizozomului conține, pe lângă proteine de transport pentru a transporta produsele de digestie în citosol, cantități mari de carbohidrați legați de lipide sau proteine ale feței necitosolice. Mai mult, se pare că lizozomul este protejat de acțiunea enzimatică a conținutului său datorită structurii tridimensionale particulare a proteinelor membranei sale interne care sunt capabile să protejeze siturile vulnerabile la atacurile enzimatice.
PH-ul scăzut este creat și menținut de pompele de protoni care pompează ioni H + din citoplasmă, precum și din canalele ionice .
Autofagie , heterofagie , autofagozom
Deoarece enzimele conținute în lizozom sunt potențial periculoase, ele nu pot fi eliberate în citoplasmă, ci trebuie întotdeauna să fie limitate în structuri membranare capabile să le conțină. Mecanismul prin care aceste enzime intră în contact cu substanțele de degradat este definit ca autofagie sau heterofagie în funcție de originea acestor substanțe.
- autofagia este procesul biologic de degradare a proteinelor celulare, prin intermediul veziculelor lizozomale care derivă din membrana reticulului endoplasmatic . [1] Închiderea acestor membrane determină formarea autofagozomului . În etapele următoare, prin fuziunea membranei autofagozomului cu cea a endozomului târziu sau lizozomului, se naște autolizozomul.
- În heterofagie , particulele mari (cum ar fi o bacterie sau un virus ) sunt încorporate (endocitate) de către celulă. De asemenea, în acest caz vezicula formată, de această dată derivând din membrana celulară ( endozom ), fuzionează cu lizozomul formând un singur complex vezicular în care conținutul său este digerat.
Putem defini definitiv lizozomul, organul care prin utilizarea enzimelor sale distruge organele în disfuncție, arde substanțele nutritive, elimină deșeurile și substanțele toxice și implementează un fel de „dosar de muncă” în celula eucariotă, distrugând deșeurile de țesuturi create în procesul de instruire. Datorită lizozomilor este de asemenea posibilă implementarea practicii apoptozei (moartea celulară programată).
Formare
Acest articol sau secțiune ar trebui revizuit și actualizat cât mai curând posibil . |
Lizozomul se formează prin înmugurirea din aparatul Golgi care procesează și enzimele litice produse de reticulul endoplasmatic. Aceste enzime sunt direcționate în lizozomi prin fosforilare la nivelul părții cis a Golgi de către o fosfotransferază care formează un reziduu de manoză -6-fosfat. Enzimele marcate cu acest motiv sunt direcționate în mod specific către pre-lizozomi (așa definite deoarece pH-ul nu este suficient de acid) prin vezicule endosomale cu pH mai mic. Prin urmare, pe măsură ce veziculele noi care transportă enzime noi se îmbină cu pre-lizozomul, pH-ul său este redus, activând în cele din urmă enzimele litice și transformându-se într-un lizozom real.
Lizozomii observați la microscopul electronic sunt prezentați sub 3 aspecte structurale, lizozomii primari, secundari și terțiari.
- Lizozomi primari: apar sub forma unor mici vezicule sferice care au un conținut ridicat de hidrolaze acide în interiorul lor. Aceste enzime lizozomale sunt sintetizate în ribozomii atașați la reticulul endoplasmatic, se acumulează în lumenul aceluiași (RE) de unde trec apoi în complexul Golgi de unde este eliberat sub formă de mici vezicule învelite de Clathrin, din fata trans .
- Lizozomi secundari (endolizozomi): se formează prin fuziunea lizozomilor primari cu endozomi, adică cu vacuoli care conțin material extracelular care a pătruns în celulă prin endocitoză. Acesta este motivul pentru care acești lizozomi se numesc endolizozomi. Endolisozomii pot funcționa cu alți lizozomi, formând un fagolizozom. Organismele celulare modificate sunt învelite și izolate cu o membrană. După aceasta, forma respectivă fuzionează cu un lizozom secundar obținând un autofagolizozom, adică o formațiune în care materialul intracelular al celulei se auto-rănește. Produsele obținute din digestia care are loc la nivelul endozomilor ajung în citoplasmă și sunt refolosite de celulă. Sunt practic reciclate.
- Lizozomi terțiari (corpuri reziduale): corpurile reziduale care rezistă procesului de digestie intracelulară sub formă de resturi nedigestibile constituie lizozomii terțiari. Unele sunt eliminate din celulă prin exocitoză, în timp ce altele sunt depuse în interiorul celulei, depozitate în granule de lipofuscină . O prezență importantă a granulelor este sinonimă cu îmbătrânirea celulară.
Relevanță clinică
Mai multe boli, foarte deseori ereditare , precum boala Tay-Sachs sau boala Pompe au fost asociate cu disfuncționalități ale lizozomilor, ale enzimelor acestora sau ale proceselor care duc la formarea lor. Aceste boli se datorează lipsei de producție a semnalului manoză-6-fosfat și lipsei unor proteine litice particulare, ceea ce duce la acumularea substraturilor lor în interiorul celulei, cu consecințele problemelor în metabolismul celular.
Până în prezent, sunt cunoscute peste 40 de boli genetice diferite cauzate de deficiența acestor enzime. Aceste boli, cunoscute în general ca Boli de stocare lizozomală sau LSD (literalmente boli de stocare lizozomală ); pot fi clasificate în grupuri în funcție de tipul de enzimă care lipsește (și, prin urmare, de tipul de produs care se acumulează):
- mucopolizaharidoză (MPS) tip I-VII, cauzată de defecte în degradarea mucopolizaharidelor (sau glicozaminoglicanilor )
- sfingolipidoză (sau glicolipidoză), în care există un defect de degradare a sfingomielelor, cerebrozidelor și gangliozidelor, componente importante ale celulelor nervoase: printre acestea se remarcă gangliozidoza GM2 ( boala Tay-Sachs )
- oligozaharidoză , în care există un defect în degradarea oligozaharidelor și glicoproteinelor
- boli datorate transportului lizozomal modificat , în care unele substanțe nu sunt transportate corect în lizozomi pentru a fi degradate
- boli datorate lipsei de transport a enzimelor lizozomale , în care unele enzime nu sunt transportate corect în lizozomi și, prin urmare, nu își îndeplinesc funcția
- alte tipuri de boli lizozomale, inclusiv boala Pompe .
Notă
Bibliografie
- Dunn WA Jr, Autofagie și mecanisme conexe ale degradării proteinelor mediate de lizozom , în Trends Cell Biol , vol. 4, nr. 4, 1994, pp. 139-43, PMID 14731737 .
- Neil Campbell și Jane Reece (2002). Biologie ed. A VI-a. Benjamin Cummings. San Francisco. ISBN 0-8053-6624-5
- Lubert Stryer (1996). Biochimie , ed. A IV-a. Zanichelli. ISBN 88-08-09806-0
Elemente conexe
linkuri externe
- Lisosoma , pe Treccani.it - Enciclopedii online , Institutul Enciclopediei Italiene .
- Lisosoma , pe Sapienza.it , De Agostini .
- ( EN ) Lysosoma , în Encyclopedia Britannica , Encyclopædia Britannica, Inc.
- ( EN ) Film flash pe lizozom , pe maxanim.com .
- ( EN ) Structuri 3D ale proteinelor asociate cu membrana lizozomală , pe opm.phar.umich.edu .
- ( EN ) Science Primer , la ncbi.nlm.nih.gov .
- boli lizozomale-Telethon , pe telethon.it (arhivat din url-ul original la 18 octombrie 2006) .
- mucopolisaccaridosi.it .
- Lisosoma , în Treccani.it - Enciclopedii online , Institutul Enciclopediei Italiene.
| Controlul autorității | Tezaur BNCF 20128 · LCCN (EN) sh85079197 · BNF (FR) cb12124912g (dată) · NDL (EN, JA) 00.569.577 |
|---|