Moleculă
În fizică și chimie , molecula (din molecula științifică latină , derivată la rândul ei din alunițe , care înseamnă „aluniță”, adică „cantitate mică”) este o entitate neutră din punct de vedere electric compusă din doi sau mai mulți atomi uniți printr-o legătură covalentă . [1] [2] În definiția Compendiului de terminologie chimică a IUPAC atomii formează o gaură de potențial Coulomb suficient de adâncă pentru a permite prezența a cel puțin unei stări vibraționale. [3]
Poate fi compus din mai mulți atomi ai aceluiași element sau din elemente diferite și identifică o substanță , din care constituie unitatea fundamentală. Moleculele formate din aceiași atomi cu un aranjament diferit în spațiu sunt numite izomeri ai unei substanțe și diferă în ceea ce privește proprietățile fizice.
În chimia organică și biochimică , termenul de moleculă identifică uneori și ioni poliatomici , în timp ce în teoria cinetică a gazelor este adesea folosit pentru fiecare particulă gazoasă, indiferent de compoziția sa: cu această definiție chiar și atomii singulari din familia gazelor nobile pot fi considerate molecule. [4]
Dinamica moleculară
Descrierea materiei la nivel atomic folosește formalismul mecanicii cuantice , care prin caracterizarea probabilistică a unei particule oferite de funcția de undă permite explicarea naturii electromagnetice a legăturilor fizice și chimice care guvernează comportamentul moleculelor și al constituenților acestora. În acest context, studiul dinamicii moleculare se bazează pe aproximarea Born-Oppenheimer , numită și aproximare adiabatică , care consideră mișcarea nucleelor independentă de cea a electronilor , deoarece primii sunt extrem de grei și, prin urmare, mai încet decât secunde. Acest lucru face posibilă factorizarea funcției de undă totală a moleculei: [5] [6]

unde indicele e indică funcția de undă a electronilor, indicele n al nucleelor și Și sunt pozițiile nucleilor și, respectiv, ale electronilor.


Această funcție de undă satisface ecuația valorii proprii :
![\ left [T_ \ mathrm {e} + T_ \ mathrm {n} + V_ \ mathrm {ne} (\ mathbf {R}, \ mathbf {r}) + V_ \ mathrm {ee} (\ mathbf {r}) + V_ \ mathrm {nn} (\ mathbf {R}) \ right] \ Psi _ {\ mathrm {total}} (\ mathbf {R}, \ mathbf {r}) = E (\ mathbf {R}) \ Psi_ {\ mathrm {total}} (\ mathbf {R}, \ mathbf {r})](https://wikimedia.org/api/rest_v1/media/math/render/svg/5fe6c950c0c256deb9804be59b253d5b0b159d0b)
unde este este energia cinetică a electronilor, cel al nucleilor, interacțiunea Coulomb între nuclei și electroni, interacțiunea Coulomb dintre electroni și acela dintre nuclei.





În aproximarea adiabatică, funcția de undă electronică este necesară pentru a satisface ecuația valorii proprii:
![\ left [T_ \ mathrm {e} + V_ \ mathrm {ne} (\ mathbf {R}, \ mathbf {r}) + V_ \ mathrm {ee} (\ mathbf {r}) \ right] \ psi _ { \ mathrm {e}} (\ mathbf {R}, \ mathbf {r}) = E _ {\ mathrm {e}} (\ mathbf {R}) \ psi _ {\ mathrm {e}} (\ mathbf { R}, \ mathbf {r})](https://wikimedia.org/api/rest_v1/media/math/render/svg/ad4ab644a0a73006f448d26d3dea2c5b0e598731)
Expresia anterioară este obținută datorită faptului că operatorul , cuprins în termen , nu acționează asupra coordonatelor nucleelor, astfel încât funcția de undă a nucleelor poate fi colectată ca un factor comun.

Funcția de undă a nucleelor, pe de altă parte, se obține pornind de la ecuația totală, care, făcând explicit operatorul de impuls, devine:
![\ left (- \ sum_ {i} {\ frac {\ hbar ^ 2} {2M_i} \ nabla_R ^ 2} + \ left [T_ \ mathrm {e} + V_ \ mathrm {ne} (\ mathbf {R}, \ mathbf {r}) + V_ \ mathrm {ee} (\ mathbf {r}) + V_ \ mathrm {nn} (\ mathbf {R}) \ right] \ right) \ psi _ {\ mathrm {e}} (\ mathbf {r}, \ mathbf {R}) \ psi _ {\ mathrm {n}} (\ mathbf {R}) = E (\ mathbf {R}) \ psi _ {\ mathrm {e}} ( \ mathbf {r}, \ mathbf {R}) \ psi _ {\ mathrm {n}} (\ mathbf {R})](https://wikimedia.org/api/rest_v1/media/math/render/svg/3bcccd002e0dc7b9a90ae914fa9145213fec19f2)
Fiind că:
![\ nabla_R ^ 2 \ left [\ psi _ {\ mathrm {e}} (\ mathbf {r}, \ mathbf {R}) \ psi _ {\ mathrm {n}} (\ mathbf {R}) \ right] = \ psi _ {\ mathrm {e}} (\ mathbf {r}, \ mathbf {R}) \ nabla_R ^ 2 \ psi _ {\ mathrm {n}} (\ mathbf {R}) + 2 \ left [ \ nabla_R \ psi_ {\ mathrm {e}} (\ mathbf {r}, \ mathbf {R}) \ right] \ nabla_R \ psi _ {\ mathrm {n}} (\ mathbf {R}) + \ psi _ {\ mathrm {n}} (\ mathbf {R}) \ nabla_R ^ 2 \ psi _ {\ mathrm {e}} (\ mathbf {r}, \ mathbf {R})](https://wikimedia.org/api/rest_v1/media/math/render/svg/2ea2510d14abcee8538b1cb4344aace0ab9e8db4)

Primesti:
![- \ sum_ {i} {\ frac {\ hbar ^ 2} {2M_i}} \ left (\ psi _ {\ mathrm {e}} (\ mathbf {r}, \ mathbf {R}) \ nabla_R ^ 2 \ psi_ {\ mathrm {n}} (\ mathbf {R}) + 2 \ left [\ nabla_R \ psi _ {\ mathrm {e}} (\ mathbf {r}, \ mathbf {R}) \ right] \ nabla_R \ psi_ {\ mathrm {n}} (\ mathbf {R}) + \ psi _ {\ mathrm {n}} (\ mathbf {R}) \ nabla_R ^ 2 \ psi _ {\ mathrm {e}} (\ mathbf {r}, \ mathbf {R}) \ right) +](https://wikimedia.org/api/rest_v1/media/math/render/svg/89ffcd328734f831bafdb86ae13f429cd577208f)
![+ \ left [T_ \ mathrm {e} + V_ \ mathrm {ne} (\ mathbf {R}, \ mathbf {r}) + V_ \ mathrm {ee} (\ mathbf {r}) + V_ \ mathrm {nn } (\ mathbf {R}) \ right] \ psi _ {\ mathrm {e}} (\ mathbf {r}, \ mathbf {R}) \ psi _ {\ mathrm {n}} (\ mathbf {R} ) = E (\ mathbf {R}) \ psi _ {\ mathrm {e}} (\ mathbf {r}, \ mathbf {R}) \ psi _ {\ mathrm {n}} (\ mathbf {R})](https://wikimedia.org/api/rest_v1/media/math/render/svg/2ca3c9a16306a86ac00cd73be598b132d0f3b6aa)
care, neglijând pentru aproximarea adiabatică termenul:
![2 \ left [\ nabla_R \ psi _ {\ mathrm {e}} (\ mathbf {r}, \ mathbf {R}) \ right] \ nabla_R \ psi _ {\ mathrm {n}} (\ mathbf {R} ) + \ psi _ {\ mathrm {n}} (\ mathbf {R}) \ nabla_R ^ 2 \ psi _ {\ mathrm {e}} (\ mathbf {r}, \ mathbf {R})](https://wikimedia.org/api/rest_v1/media/math/render/svg/802313832f8fd89f3dc299d9b4208fd68370a527)
devine, prin introducerea soluției ecuației electronice:

![\ left [- \ sum_ {i} {\ frac {\ hbar ^ 2} {2M_i}} \ nabla_R ^ 2 + E _ {\ mathrm {e}} (\ mathbf {R}) + V_ \ mathrm {nn} (\ mathbf {R}) \ right] \ psi _ {\ mathrm {n}} (\ mathbf {R}) = E _ {\ mathrm {n}} \ psi _ {\ mathrm {n}} (\ mathbf {R})](https://wikimedia.org/api/rest_v1/media/math/render/svg/cd9d24946969a24f4827c1eee728265335cda01c)
care este ecuația mișcării nucleelor.
Potențialul care conduce mișcarea nucleelor:

se numește potențial adiabatic sau potențial intermolecular și stă la baza dinamicii moleculei.
Din expresia potențialului adiabatic este clar că dinamica nucleelor este condusă de energie furnizat de ecuația electronică: acest termen este fundamental, deoarece reprezintă „lipiciul” care ține împreună nucleii atomilor care alcătuiesc molecula. [7]
Pentru moleculele diatomice, potențialul adiabatic este un potențial armonic și poate fi aproximat de potențialul Morse , care, spre deosebire de oscilatorul armonic cuantic, include în mod explicit efectele ruperii legăturilor chimice , cum ar fi existența stărilor nelegate.
Molecule diatomice
Moleculele diatomice sunt compuse din doi atomi și se disting în molecule homonucleare, atunci când atomii sunt din același element chimic , și heteronucleari, când atomii diferă.
Molecula H 2 +


Moleculele diatomice homonucleare sunt compuse din doi atomi ai aceluiași element chimic; cea mai simplă dintre acestea este H 2 + , pentru care ecuația electronică ia forma: [8]
![\ left [{\ frac {\ hbar ^ 2} {2m_e}} \ nabla_r ^ 2 - \ frac {ke ^ 2} {| \ mathbf {r} + \ mathbf {R} / 2 |} - \ frac {ke ^ 2} {| \ mathbf {r} - \ mathbf {R} / 2 |} + \ frac {ke ^ 2} {R} \ right] \ psi _ {\ mathrm {e}} (\ mathbf {r} ) = E _ {\ mathrm {e}} (\ mathbf {r}) \ psi _ {\ mathrm {e}} (\ mathbf {r})](https://wikimedia.org/api/rest_v1/media/math/render/svg/dbdcc15a178e5f1487ae8efb1746b1a94d4898bc)
unde este , al doilea și al treilea termen reprezintă atracția V ne a electronului către nuclei și al patrulea repulsia celor doi nuclei.
Cei doi protoni formează două puțuri potențiale, iar funcția de undă electronică este combinația liniară a două funcții de undă asemănătoare hidrogenului : [9]


![\ psi _ {\ mathrm {\ pm}} (\ mathbf {r}) = \ frac {1} {\ sqrt {2}} [\ psi_ {1s} (\ mathbf {r} + \ mathbf {R} / 2) \ pm \ psi_ {1s} (\ mathbf {r} - \ mathbf {R} / 2)]](https://wikimedia.org/api/rest_v1/media/math/render/svg/fdf6c56809c63e4c6bfd05f1afb629af6a11f059)
Funcția de undă constituie legătura moleculară orbitală , funcția constituie orbitalul anti - legătură . [10] Orbitalul de legare are o energie mai mică decât orbitalul anti-legătură.
Funcții , deși descriu bine distribuția probabilității electronului în starea fundamentală, nu sunt soluții exacte ale ecuației electronice.



Funcția de undă , în spațiul dintre cele două nuclee, este mai mare decât funcțiile individuale de undă asemănătoare hidrogenului , și acest fapt generează legătura covalentă între cei doi nuclei. De fapt, se observă că densitatea de probabilitate asociată cu funcția de undă:

![| \ psi _ {\ mathrm {\ pm}} | ^ 2 = \ frac {1} {2} [\ psi_ {1s} ^ 2 (\ mathbf {r} + \ mathbf {R} / 2) + \ psi_ {1s} ^ 2 (\ mathbf {r} - \ mathbf {R} / 2) \ pm 2 \ psi_ {1s} (\ mathbf {r} + \ mathbf {R} / 2) \ psi_ {1s} (\ mathbf {r} - \ mathbf {R} / 2)]](https://wikimedia.org/api/rest_v1/media/math/render/svg/50a75eda255a84496516016b11905999d90085a1)
conține un termen de interacțiune, produsul dublu, care reprezintă suprapunerea celor două funcții de undă: este o regiune de sarcină negativă care unește cei doi nuclei de sarcină opusă.
În ceea ce privește orbitalul anti-legătură , dispare la mijloc între cele două nuclee, unde generează o densitate de probabilitate mai mică decât ar avea fără termenul de suprapunere.
Molecula H 2


Acum luați în considerare molecula H 2 , cea mai simplă moleculă neutră. Având doi electroni, funcția de undă electronică singlet este dată de: [11]
![\ psi _ {\ mathrm {S}} (1,2) = \ frac {1} {\ sqrt {2}} [\ psi_ {1s} (\ mathbf {r_1} - \ mathbf {R} / 2) \ psi_ {1s} (\ mathbf {r_2} + \ mathbf {R} / 2) + \ psi_ {1s} (\ mathbf {r_2} - \ mathbf {R} / 2) \ psi_ {1s} (\ mathbf {r_1 } + \ mathbf {R} / 2)] \ chi ^ A (1,2)](https://wikimedia.org/api/rest_v1/media/math/render/svg/909792d0f57537b94dea0a27378c910077e1d41c)
și reprezintă legătura orbitală, în timp ce cea a tripletului din:[12]
![\ psi _ {\ mathrm {T}} (1,2) = \ frac {1} {\ sqrt {2}} [\ psi_ {1s} (\ mathbf {r_1} - \ mathbf {R} / 2) \ psi_ {1s} (\ mathbf {r_2} + \ mathbf {R} / 2) - \ psi_ {1s} (\ mathbf {r_2} - \ mathbf {R} / 2) \ psi_ {1s} (\ mathbf {r_1 } + \ mathbf {R} / 2)] \ chi ^ S (1,2)](https://wikimedia.org/api/rest_v1/media/math/render/svg/efa707e0f683775b3cc7ef364bdf94eb4370d291)
reprezentând orbitalul anti-legătură, unde:

Și

sunt stările de rotire , unde + reprezintă rotirea, - rotirea.
Densitatea probabilității spațiale este:[12]

![\ pm 2 \ psi_ {1s} (\ mathbf {r_1} - \ mathbf {R} / 2) \ psi_ {1s} (\ mathbf {r_1} + \ mathbf {R} / 2) \ psi_ {1s} (\ mathbf {r_2} - \ mathbf {R} / 2) \ psi_ {1s} (\ mathbf {r_2} + \ mathbf {R} / 2)]](https://wikimedia.org/api/rest_v1/media/math/render/svg/acbf4078b38a04bc2233a417ca8382bb3bc4f732)
De asemenea, în acest caz, termenul de interferență reprezintă suprapunerea funcțiilor de undă de tip hidrogen în regiunea dintre nuclee și implică o creștere a sarcinii în cazul singletului (semnul +) și o scădere a sarcinii în triplet (- semn).
Molecule heteronucleare
La moleculele heteronucleare simetria care a caracterizat moleculele homonucleare lipsește, iar orbitalele nu sunt o combinație pură simetrică și antisimetrică a orbitalilor atomici. În astfel de molecule, orbitalele pot fi aproximate cu stările proprii ale unei matrici pătrate de dimensiunea 2: [13]

unde este:

este hamiltonianul eficient al electronului unic în timp ce stările Și sunt orbitalele corespunzătoare atomului stâng și respectiv al celui drept.
Valorile proprii asociate matricei sunt:



Orbitalii de legare și anti-legare sunt date de statele proprii:



cu:

pentru obținem molecula homonucleară și termenul reprezintă împărțirea între orbitalul de legare și antiblocarea unei molecule homonucleare sau împărțirea între combinații simetrice și antisimetrice. [13]
Dupa cum statele proprii de legare și anti-legătură seamănă din ce în ce mai mult cu orbitalii Și a atomilor individuali și același lucru se întâmplă pentru valorile proprii energetice respective. [14] Când diferența este de așa natură încât implică un transfer complet de sarcină între cei doi atomi, legătura se spune că este ionică .







Molecule polatomice
Moleculele poliatomice au mai mult de doi atomi, care în majoritatea cazurilor sunt diferite între ele. Structura lor este extrem de diversă, deoarece posibilele combinații între orbitalii atomici care formează orbitalii moleculari sunt extrem de numeroși.
Pe lângă legătura care caracterizează moleculele diatomice, în moleculele poliatomice orbitalele atomice s și p pot fi combinate între ele pentru a forma orbitalele numite hibrizi .
Două exemple de molecule poliatomice sunt raportate mai jos, apă și metan :
Molecula H 2 O
Una dintre cele mai simple molecule poliatomice este cea a apei , în care oxigenul are un orbital p caracterizat printr-o triplă degenerare pe cele trei axe carteziene, care generează două configurații electronice posibile: prima este cazul în care cei 4 electroni umplu complet doi lobi a orbitalului, lăsându-l pe al treilea gol, în timp ce al doilea este cazul în care există doi electroni pe un lob și unul pe fiecare dintre cei doi rămași. Prin urmare, acest orbital poate fi scris ca 2 p x p y p z 2 , în care se presupune că lobul îndreptat de-a lungul axei z conține doi electroni și acest lucru face posibilă formarea a două legături covalente, în care lobii x și y leagă cei doi atomi de hidrogen . [15]
Molecula CH 4
Metanul este o moleculă cu un orbital hibrid. Carbonul are o configurație electronică 1 s 2 2 s 2 2 p 2 , iar orbitalul p e în starea sa de bază se poate lega, prin urmare, cu doar doi atomi de hidrogen. Molecula de metan există, totuși, deoarece un electron din orbitalul de 2 s 2 este promovat în orbitalul p , astfel încât configurația electronică devine 1 s 2 2 s 2 p x p y p z , generând patru electroni decuplați care se pot lega de la fel de mulți atomi de hidrogen.
Cei patru orbitali moleculari hibrizi sunt, prin urmare, o combinație liniară a stărilor , , , a formei: [16]





și formează un tetraedru cu atomul de carbon în centru.
Orbitali și legături moleculare
Orbitalul molecular caracterizează configurația electronică a unei molecule, definind distribuția spațială și energia electronilor și a fost introdus de Friedrich Hund [17] [18] și Robert S. Mulliken [19] [20] în 1927 și 1928. [21 ] [22]
Un orbital molecular este reprezentat de o funcție de undă al cărei pătrat descrie distribuția probabilității în raport cu poziția electronului. Această funcție de undă este obținută din ecuația de undă care descrie întreaga moleculă, ceea ce în general nu este ușor de rezolvat: această problemă este rezolvată prin intermediul unei aproximări care constă în scrierea orbitalului molecular ca o combinație liniară a orbitalilor individului. atomi. Această aproximare este descrisă de teoria orbitalilor moleculare .
Ordinea legăturii este, de asemenea, semidiferența dintre numărul de electroni de legare și numărul de electroni anti-legătură. Ordinea de obligațiuni este un indice al puterii legăturii în sine și este , de asemenea , utilizat pe scară largă în teoria legătură de valență .
Teoria orbitală moleculară
Teoria orbitalilor moleculari este o tehnică pentru determinarea structurii moleculare în care electronii nu sunt repartizați la anumite legături chimice, ci sunt tratați ca obiecte care se mișcă sub influența nucleelor din întreaga moleculă. [23]

Funcția de undă totală a electronilor este scris ca o combinație liniară : [24]


unde este sunt orbitalii atomici și coeficienții însumare, obținute prin rezolvarea ecuației Schrödinger pentru și aplicarea principiului variațional .
Principalele proprietăți ale orbitalilor moleculari astfel definiți sunt:



- Numărul orbitalilor moleculari este egal cu numărul orbitalilor atomici conținuți în combinația liniară din care sunt făcuți, deoarece stările staționare nu sunt nici create, nici distruse. [25]
- Dacă molecula posedă simetrii, orbitalii atomici degenerate, caracterizate prin aceeași energie, sunt grupate în combinații liniare ce aparțin reprezentarea a simetrie grupului .
- Numărul orbitalilor moleculari care aparțin reprezentării unui grup este egal cu numărul orbitalilor atomici care aparțin acelei reprezentări.
- Într-o anumită reprezentare, orbitalii atomici se amestecă mai mult cu cât nivelurile lor de energie atomică sunt mai apropiate.
Reprezentarea orbitalilor moleculari
Nomenclatorul orbitali moleculare urmeaza cea a orbital atomic: când un orbital are o simetrie cilindrică în ceea ce privește îmbinarea celor două nuclee numită direcția de obligațiuni, este indicat cu litera grecească ; când este pe laturile opuse în raport cu direcția de legare cu care este indicat . Alături de literă este scris un index care indică din ce tip de legătură atomică se formează orbitalul molecular. [26]
Există, de asemenea, un al treilea tip de obligațiune, notat cu , obținută prin suprapunerea a patru lobi a doi orbitali atomici. În acest caz , există două planuri nodale situate între cele două nuclee care contractul această legătură. Legătura δ se găsește în legătura cvadruplă , o legătură multiplă importantă în chimia anorganică și care caracterizează complexe precum [Re 2 Cl 10 ] 4- sau alte tipuri de clustere .



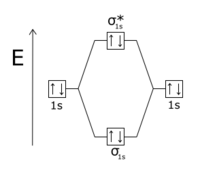
Orbitalul anti-legătură este, de asemenea, notat printr-un asterisc, de exemplu, molecula H 2 are un orbital de legătură și un orbital anti-legătură .


- În moleculele diatomice homonucleare, electronii umple orbitalii cu același model cu care are loc umplerea orbitalilor atomici, cu singura excepție că dintre orbitalii care derivă din orbitalii atomici de 2 p , orbitalii , au energie mai mică decât orbitalii datorită faptului că repulsia Coulomb a orbitalelor derivate din orbitalele atomice de 1 s și 2 s mărește energia stărilor . Acest lucru se datorează faptului că electronii celor două legături acestea sunt situate în regiunea dintre cele două nuclee și, prin urmare, se resping reciproc; orbitali în molecule mai grele decât oxigenul au mai puțină energie și sunt situate în apropierea nucleelor, de aceea ordinea energetică naturală este restabilită.
Combinația liniară a funcțiilor de undă care formează orbitalul molecular este prezentată pe lateral, unde molecula He 2 și molecula O 2 sunt schematizate, care are o configurație electronică: . [27] - Nel caso di molecole biatomiche eteronucleari, se il numero atomico dei due atomi differisce di poco il procedimento che forma gli orbitali è lo stesso delle molecole omonucleari. Vi è tuttavia una differenza di elettronegatività tra i due atomi, e ciò implica la presenza di un dipolo elettrico tra di essi dovuto al fatto che gli elettroni si distribuiscono nelle vicinanze dell'atomo più elettronegativo: [28] il legame che si viene a formare prende il nome di covalente polare .
 La molecola del monossido di carbonio CO
La molecola del monossido di carbonio CO



Tale legame viene rappresentato come in figura a lato, e si può notare che gli elettroni di hanno energia maggiore, e costituiscono un orbitale detto HOMO (Highest Occupied Molecular Orbital), mentre gli elettroni di e costituiscono gli orbitali vuoti a minore energia detti LUMO (Lowest Unoccupied Molecular Orbital). L'orbitale LUMO è il centro in cui la molecola può subire un attacco nucleofilo di una base di Lewis , e si tratta quindi del centro di acidità di Lewis. Viceversa, HOMO è il centro di basicità di Lewis della molecola, e può subire un attacco elettrofilo.
Se la differenza di elettronegatività è maggiore di un valore convenzionale fissato a 1,9 vi è un trasferimento completo di carica tra i due atomi, cioè la nuvola elettronica può considerarsi come spostata completamente sull'elemento più elettronegativo. Tale legame prende il nome di legame ionico .
Se il numero atomico dei due atomi differisce di molto accade che gli orbitali molecolari si formino tra orbitali atomici con energia simile, invece che dello stesso tipo. [29]



- All'aumentare del numero di atomi coinvolti diventa complessa la caratterizzazione degli orbitali, a nell'ambito della teoria degli orbitali molecolari sono stati sviluppati diversi metodi di calcolo degli orbitali, tra i quali vi sono il Metodo di Hückel , proposto da Erich Hückel nel 1930 , consiste in un semplice metodo LCAO utilizzato per la determinazione delle energie degli orbitali molecolari di sistemi π rappresentati da idrocarburi con legami coniugati, risultando applicabile a molecole quali ad esempio l' etilene , il benzene e il butadiene . [30] [31] La nota regola di Hückel trae origine da queste basi.
Il metodo di Hückel esteso , sviluppato da Roald Hoffmann , rappresenta invece la base delle regole di Woodward-Hoffmann [32] ed è un'estensione a tutti gli orbitali di valenza . Negli anni successivi il metodo fu reso applicabile anche agli eterocicli come la piridina , il pirrolo e il furano . [33]
Vi è infine il metodo di Pariser–Parr–Pople , che sfrutta metodi semi-empirici della chimica quantistica nell'ambito della chimica organica .
Moti interni nelle molecole biatomiche
I nuclei sono soggetti al potenziale adiabatico definito in precedenza, che nelle molecole biatomiche è indipendente dalla posizione del centro di massa della molecola e dall'orientazione della retta congiungente i due nuclei. Il potenziale gode quindi di invarianza rispetto alle traslazioni ed alle rotazioni, e il moto dei nuclei può essere studiato come un problema a due corpi , sicché l' equazione di Schrödinger può essere separata in moto radiale, dipendente dalla distanza tra i due nuclei, e moto orbitale, dipendente dal numero quantico orbitale . L'equazione di Schrödinger nel caso di un moto in un campo centrale è:
![\left[ -\frac{\hbar^2}{2 (M+m)} \nabla_{\mathbf r_{cm}}^{2} - \frac{\hbar^2}{2\mu} \nabla^{2} + V_\mathrm{ad}(|\mathbf r_{rel}|) \right] \psi_{\mathrm{n}}(\mathbf r_{cm},\mathbf r_{rel}) = E_{tot} \psi_{\mathrm{n}}(\mathbf r_{cm},\mathbf r_{rel})](https://wikimedia.org/api/rest_v1/media/math/render/svg/2f63a966a7e40307058884854406f9068bf7029e)
dove indica la posizione del centro di massa e la posizione relativa dei due nuclei, differenza delle rispettive posizioni.
Il problema può essere quindi separato in due equazioni, una per il centro di massa ed una per la particella di massa μ che si muove in un campo centrale rispetto al centro di massa. La funzione d'onda si può quindi fattorizzare nel seguente modo: . L'equazione per , che rappresenta il problema della particella libera , fornisce l'energia traslazionale della molecola. L'equazione per si può ulteriormente fattorizzare in parte radiale, dipendente da r , e parte angolare, dipendente dalle coordinate angolari: .
La soluzione per sono le armoniche sferiche , ed i rispettivi stati sono autostati del momento angolare orbitale e della sua componente lungo l'asse z .
L'equazione per è invece, detta : [34]









![\left[ -\frac{\hbar^2}{2 \mu} \frac{d^2}{d r^2} + \frac{\hbar^2 l(l+1)}{2 \mu r^2} + V_\mathrm{ad}(|\mathbf r_{rel}|) \right] g = E g](https://wikimedia.org/api/rest_v1/media/math/render/svg/a0d8b7c5343baef9266e16f45363c9237debc159)
dove il secondo termine rappresenta il contributo energetico rotazionale , che dipende dal numero quantico orbitale l .
Il potenziale adiabatico può essere inoltre sviluppato in serie di Taylor , che troncata al secondo ordine è: [6]


dove è il valore di che minimizza , e rappresenta la posizione di equilibrio dei due nuclei. Tale espressione rappresenta un moto armonico attorno a che fornisce un contributo energetico dato dall'energia dell'equazione elettronica contenuta in e dall'energia vibrazionale .






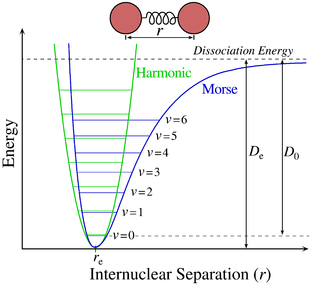
Detta la lunghezza caratteristica data dalla relazione e detta , le soluzioni dell'equazione per sono:





dove è il polinomio di Hermite di grado .
Lo spettro energetico contiene in definitiva tre termini:



Tali termini sono i contributi energetici che caratterizzano la dinamica della molecola biatomica, e nello specifico sono: [6] [35]
- Il contributo elettronico, dato dal termine di , definisce la profondità della buca di potenziale generata dai due nuclei, responsabile del legame chimico. I livelli energetici associati a questo termine sono detti superfici adiabatiche , e corrispondono ai diversi stati energetici degli elettroni. Gli elettroni che vengono promossi da un orbitale ad un altro, ad esempio da un orbitale di legame ad uno di antilegame, effettuano una transizione tra due valori e del potenziale adiabatico. Tali transizioni sono dell'ordine di 10 eV , ea differenti superfici adiabatiche corrispondono anche diversi valori di . Le transizioni elettroniche tra due di tali superfici sono inoltre accompagnate da transizioni tra diversi stati vibrazionali e rotazionali.
- Il contributo vibrazionale, meno energetico del precedente, nell'approssimazione di moto armonico fornita dall'esclusione dei termini superiori al secondo ordine nel precedente sviluppo di è dato dagli autovalori dell' oscillatore armonico quantistico :






- dove è la costante di Planck e la frequenza angolare dell'oscillazione intorno a .
- La frequenza è data da:



- con

- e la massa ridotta dell'oscillatore a due corpi, data dal rapporto tra il prodotto e la somma delle masse dei due nuclei.
- Tale contributo descrive il moto armonico dei due nuclei intorno alla posizione di equilibrio, e transizioni tra due livelli vibrazionali sono dell'ordine del decimo di eV.

- Il contributo rotazionale, il meno energetico dei tre, è fornito dall'equazione angolare dell' atomo di idrogeno , pari a:

- dove è il momento angolare orbitale e il momento d'inerzia .
- Tale contributo è generalmente dell'ordine dei meV, ed è calcolato assumendo .



In conclusione, quindi, l'energia interna di una molecola biatomica è:

dove i termini sono elencati in ordine di importanza.
Moti interni nelle molecole poliatomiche
Nelle molecole poliatomiche il calcolo dello spettro energetico può essere molto complesso. Le simmetrie della molecola giocano spesso un ruolo determinante al fine di ottenere gli autovalori dell'energia vibrazionale e rotazionale.
Moto vibrazionale
Nelle molecole poliatomiche l'energia cinetica data dal moto vibrazionale è espressa come:

dove le coordinate cartesiane sono le posizioni del nucleo α-esimo rispetto alla posizione di equilibrio.
Utilizzando coordinate mass–weighted :

è possibile definire la matrice di elementi:


E quindi, come nelle molecole biatomiche, l'energia vibrazionale può essere espressa come:

dove è il vettore che ha per componenti Le equazioni del moto sono date dal sistema di equazioni differenziali:



Ogni atomo vibra con la stessa frequenza angolare, e tali frequenze sono dette modi normali di vibrazione , che si ottengono dalle radici dell'equazione caratteristica per la matrice :

Moto rotazionale
Considerando la molecola un corpo rigido, è possibile definire il momento d'inerzia attorno a un asse a come:

Gli assi d'inerzia di una molecola sono tre, ei rispettivi momenti d'inerzia sono , , .
Se , il corpo rigido è detto asymmetrical top , se è detto symmetrical top , mentre se è detto spherical top . All'interno dei corpi rigidi symmetrical top , se il corpo è detto oblato , si tratta di una molecola piatta, come il benzene , se invece è detto prolato , e si tratta di una molecola allungata, come il pentacloruro di fosforo .
L'energia cinetica è data da:









dove , ed sono le tre componenti dell'operatore momento angolare totale di rotazione della molecola lungo gli assi di inerzia a , b e c .



- Nel caso di uno spherical top si ottiene immediatamente che gli autovalori dell'energia rotazionale sono:

- e la degenerazione degli autovalori è .

- Nel caso di un symmetrical top si ha:

- e dal momento che commuta con ogni sua componente e con , l'autofunzione associata all'energia vibrazionale è simultanea a questi tre operatori.
- L'energia rotazionale è data allora da:



- con degenerazione se m è diverso da zero, se è invece nullo.


- Il caso di asymmetrical top è più complesso, ed è necessario diagonalizzare la matrice di nella base delle autofunzioni di L e L z .

Spettro elettromagnetico molecolare




Lo spettro elettromagnetico molecolare è generato dalle transizioni tra due autostati dell'energia totale. Nel caso si studi lo spettro di emissione la molecola passa da uno stato eccitato allo stato fondamentale, mentre nel caso si studi lo spettro di assorbimento si osserva la transizione inversa. Tale passaggio comporta l'emissione o l'assorbimento di un fotone , la cui frequenza è data dalla legge di Planck :

dove è la differenza di energia tra i due stati di partenza e arrivo:


Le transizioni elettroniche dallo stato fondamentale ai primi stati eccitati sono dell'ordine di alcuni eV , e sono osservate nella regione del visibile e dell' ultravioletto dello spettro elettromagnetico , mentre le transizioni roto-vibrazionali sono osservate nella regione dell' infrarosso . [36]
Le transizioni tra due autostati dell'energia totale vengono studiate attraverso le transizioni tra autostati delmomento di dipolo elettrico , definito come: [6]

con e la carica dell'elettrone.
Tale operatore è esplicitato dall'espressione:
![\mathbf{d} = \int {\psi_{vib}'^ * \psi_{rot}'^ *} \left[\int \psi_{el}^ * \mathbf{d} \psi_{el} dx_e \right] \psi_{vib} \psi_{rot} d\tau = \langle {\psi_{vib}' \psi_{rot}'} | \mathbf{\mu} | \psi_{vib} \psi_{rot} \rangle](https://wikimedia.org/api/rest_v1/media/math/render/svg/16e3a71f413c36ee9c61f3107e06dcdcd39d8a6a)
dove è l'operatore di momento dipolare elettronico della molecola:


Ognuno dei livelli vibrazionali che caratterizzano una superficie adiabatica è associato a diversi stati rotazionali. Nel diagramma spettroscopico le transizioni rotazionali costituiscono due rami: il primo è detto R Branch , e rappresenta le transizioni rotazionali tra i numeri quantici , mentre il secondo, detto P branch , rappresenta le transizioni . Tra i due rami vi è un vuoto, motivato dal fatto che la transizione è proibita dalle regole di selezione. [37]



Quando la transizione viene effettuata da un elettrone, essa genera anche transizioni tra autostati dell'energia roto-vibrazionale dei nuclei: tali transizioni sono dette vibroniche , e sono causate dal fatto che a due differenti superfici adiabatiche corrispondono geometrie diverse della molecola. In particolare, nelle molecole biatomiche, corrispondono a distanze internucleari differenti.
Spettro nucleare

Spettro nelle molecole biatomiche
Nel caso di molecole biatomiche omonucleari il momento di dipolo elettrico è nullo per motivi di simmetria, [38] e questo fatto spiega la trasparenza dell' atmosfera terrestre , composta prevalentemente da O 2 e N 2 .
Nelle molecole biatomiche eteronucleari, invece, l'elemento di matrice della componente lungo l'asse z del momento di dipolo è: [6]


dove sono gli autostati simultanei dell'energia vibrazionale e rotazionale. Lo stesso accade per le componenti x e y .
Dalle proprietà delle armoniche sferiche e dallo sviluppo di attorno alla distanza di equilibrio si ottengono le regole di selezione:



che definiscono le transizione permesse tra autostati dell'operatore associato all' osservabile dipolo elettrico.
Spettro nelle molecole poliatomiche
L'operatore di momento dipolare elettronico di una molecola poliatomica è dato da: [6]

in cui sono i versori degli assi d'inerzia.
Il momento di dipolo elettrico diventa:


Detto il vettore delle coordinate normali , le cui componenti sono:


ed espandendo in serie di Taylor attorno alla posizione di equilibrio:


si ottengono i due termini che generano le transizioni. Le transizioni dovute al primo termine del secondo membro sono nella regione delle microonde dello spettro, mentre le transizioni dovute al secondo termine nell' infrarosso . Il secondo termine fornisce inoltre le regole di selezione relative all'oscillatore armonico corrispondente: .

Per quanto riguarda lo spettro rotazionale, si ha che gli spherical top ed i symmetrical top planari hanno dipolo nullo, e pertanto non generano transizioni di dipolo. Nel caso di symmetrical top non planari, il dipolo è diretto lungo l'asse di simmetria, e le transizioni tra autostati degli operatori , ed sono rispettivamente:


e si rilevano nella regione delle microonde dello spettro.

Spettro elettronico
Una transizione elettronica molecolare consiste in una transizione da parte dell'elettrone tra due superfici adiabatiche . Tali transizioni sono simili a quelle atomiche, e consistono nella promozione di un elettrone da un orbitale molecolare ad un altro orbitale vuoto. [36]
Le regole di selezione si ricavano osservando che l'operatore di spin totale:

commuta con l'hamiltoniana elettronica e con , l'operatore di dipolo non agisce sullo spin, e pertanto si ha che . [6]
Per l'operatore di momento angolare nelle molecole biatomiche:



solo la componente lungo l'asse z commuta con , ottenendo che , mentre per le altre due componenti si ricava che . In definitiva si ha:




Il principio di Franck Condon
Il principio di Franck Condon afferma la probabilità associata ad una transizione vibrazionale, data da:

aumenta all'aumentare della sovrapposizione delle funzioni d'onda dei rispettivi stati iniziale e finale. Questo comporta che i livelli vibrazionali associati allo stato finale sono favoriti nel momento in cui la transizione comporta un cambiamento minimo nelle coordinate nucleari. Una conseguenza del principio è che, ad esempio, come mostrato nella figura a sinistra, se le funzioni d'onda tra lo stato fondamentale della superficie adiabatica iniziale e il secondo stato eccitato della superficie adiabatica finale si sovrappongono, tale transizione è più probabile delle altre dal momento che minimizza la variazione delle coordinate dei nuclei.
Note
- ^ Pauling, Linus .
- ^ Ebbin, Darrell .
- ^ AD McNaught, A. Wilkinson, IUPAC. Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Versione online corretta: (2006) , su goldbook.iupac.org , Blackwell Scientific Publications, Oxford.
- ^ Sulekh Chandra, Comprehensive Inorganic Chemistry , New Age Publishers, ISBN 81-224-1512-1 .
- ^ Manini , Pag. 61 .
- ^ a b c d e f g Renzo Cimiraglia - Note al corso di Spettroscopia Molecolare ( PDF ), su chim183.unife.it . URL consultato il 15 novembre 2010 (archiviato dall' url originale il 2 agosto 2007) .
- ^ Manini , Pag. 62 .
- ^ Brehm, Mullins , Pag. 503 .
- ^ Brehm, Mullins , Pag. 504 .
- ^ Brehm, Mullins , Pag. 507 .
- ^ Brehm, Mullins , Pag. 509 .
- ^ a b Brehm, Mullins , Pag. 510 .
- ^ a b Manini , Pag. 70 .
- ^ Manini , Pag. 71 .
- ^ Brehm, Mullins , Pag. 521 .
- ^ Brehm, Mullins , Pag. 522 .
- ^ F. Hund, "Zur Deutung einiger Erscheinungen in den Molekelspektren" [On the interpretation of some phenomena in molecular spectra] Zeitschrift für Physik , vol. 36, pages 657-674 (1926).
- ^ F. Hund, "Zur Deutung der Molekelspektren," Zeitschrift für Physik , Part I, vol. 40, pages 742-764 (1927); Part II, vol. 42, pages 93-120 (1927); Part III, vol. 43, pages 805-826 (1927); Part IV, vol. 51, pages 759-795 (1928); Part V, vol. 63, pages 719-751 (1930).
- ^ RS Mulliken, "Electronic states. IV. Hund's theory; second positive nitrogen and Swan bands; alternate intensities," Physical Review , vol. 29, pages 637 - 649 (1927).
- ^ RS Mulliken, "The assignment of quantum numbers for electrons in molecules," Physical Review , vol. 32, pages 186 - 222 (1928).
- ^ Friedrich Hund and Chemistry, Werner Kutzelnigg, on the occasion of Hund's 100th birthday, Angewandte Chemie , 35, 573 - 586, (1996)
- ^ Robert S. Mulliken 's Nobel Lecture, Science , 157, no. 3785, 13 - 24, (1967).
- ^ Daintith, J., Oxford Dictionary of Chemistry , New York, Oxford University Press, 2004, ISBN 0-19-860918-3 .
- ^ Licker, Mark, J., McGraw-Hill Concise Encyclopedia of Chemistry , New York, McGraw-Hill, 2004, ISBN 0-07-143953-6 .
- ^ Spinicci , Pag. 185 .
- ^ Spinicci , Pag. 181 .
- ^ Spinicci , Pag. 182 .
- ^ Spinicci , Pag. 187 .
- ^ Spinicci , Pag. 188 .
- ^ E. Hückel, Zeitschrift für Physik , 70, 204, (1931); 72, 310, (1931); 76, 628 (1932); 83, 632, (1933)
- ^ Hückel Theory for Organic Chemists , CA Coulson, B. O'Leary and RB Mallion, Academic Press, 1978
- ^ Stereochemistry of Electrocyclic Reactions RB Woodward, Roald Hoffmann J. Am. Chem. Soc.; 1965; 87(2); 395-397
- ^ Andrew Streitwieser, Molecular Orbital Theory for Organic Chemists , Wiley, New York, 1961
- ^ Brehm, Mullins , Pag. 523 .
- ^ Manini , Pag. 76 .
- ^ a b Manini , Pag. 79 .
- ^ Manini , Pag. 78 .
- ^ Brehm, Mullins , Pag. 528 .
Bibliografia
- ( EN ) John Brehm, William J. Mullins,Introduction To The Structure Of Matter: A Course In Modern Physics , Greenville, NC, USA, John Wiley & Sons, 1989, ISBN 978-0-471-60531-7 .
- ( EN ) Nicola Manini, Introduction to the Physics of Matter , Milano, CUSL, 2008, ISBN 978-88-8132-552-8 .
- Roberto Spinicci, Elementi di Chimica , Firenze, Firenze University Press, 2009, ISBN 978-88-6453-062-8 .
- ( EN ) Pauling, Linus, General Chemistry , New York, Dover Publications, Inc., 1970, ISBN 0-486-65622-5 .
- ( EN ) Ebbin, Darrell, D., General Chemistry, 3rd Ed. , Boston, Houghton Mifflin Co., 1990, ISBN 0-395-43302-9 .
- ( EN ) Brown, TL, Chemistry – the Central Science, 9th Ed. , New Jersey, Prentice Hall, 2003, ISBN 0-13-066997-0 .
- ( EN ) Chang, Raymond, Chemistry, 6th Ed. , New York, McGraw Hill, 1998, ISBN 0-07-115221-0 .
- ( EN ) Zumdahl, Steven S., Chemistry, 4th ed. , Boston, Houghton Mifflin, 1997, ISBN 0-669-41794-7 .
Voci correlate
- Atomo
- Composto organico
- Formula chimica
- Macromolecola
- Molecola biatomica
- Interazione debole
- Isomeria
- Legame chimico
- Simmetria molecolare
- Storia della chimica
- Orbitale molecolare
Altri progetti
-
 Wikizionario contiene il lemma di dizionario « molecola »
Wikizionario contiene il lemma di dizionario « molecola » -
 Wikimedia Commons contiene immagini o altri file sulla molecola
Wikimedia Commons contiene immagini o altri file sulla molecola
Collegamenti esterni
- Molecola , su Treccani.it – Enciclopedie on line , Istituto dell'Enciclopedia Italiana .
- Molecola / Molecola (altra versione) , in Enciclopedia Italiana , Istituto dell'Enciclopedia Italiana .
- ( EN ) Molecola , su Enciclopedia Britannica , Encyclopædia Britannica, Inc.
- Molecola , in Dizionario delle scienze fisiche , Istituto dell'Enciclopedia Italiana, 1996.
- Molecola , in Enciclopedia della scienza e della tecnica , Istituto dell'Enciclopedia Italiana, 2007-2008.
- Molecola , in Enciclopedia dei ragazzi , Istituto dell'Enciclopedia Italiana, 2005-2006.
| Controllo di autorità | Thesaurus BNCF 4215 · LCCN ( EN ) sh85086597 · GND ( DE ) 4039972-2 · BNF ( FR ) cb119469207 (data) · BNE ( ES ) XX524812 (data) · NDL ( EN , JA ) 00561032 |
|---|