Sindromul Klinefelter
| Sindromul Klinefelter | |
|---|---|
 | |
| Specialitate | genetică clinică |
| Clasificare și resurse externe (EN) | |
| Plasă | D007713 |
| MedlinePlus | 000382 |
| eMedicină | 945649 |
| Sinonime | |
| Sindromul Klinefelter-Reifenstein-Albright | |
| Eponime | |
| Harry Fitch Klinefelter Jr. | |
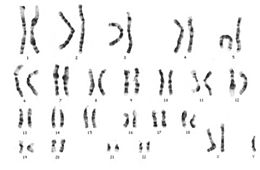
Sindromul Klinefelter este o tulburare genetică cronică caracterizată printr-o anomalie cromozomială în care un individ masculin are un cromozom X supranumerar. În mod normal, femeile au doi cromozomi sexuali XX și bărbații unul X și unul Y : indivizii cu sindrom Klinefelter au cel puțin doi cromozomi X și cel puțin un cromozom Y[1] . Prin urmare, persoanele cu acest cariotip sunt denumite de obicei „bărbați XXY” sau „47, XXY”[2] . Cu toate acestea, un corp Barr este prezent, altfel absent la bărbați.
Această afecțiune apare la aproximativ 1-2 bărbați din 1.000 de nașteri vii [3] [4] . Mulți oameni cu sindrom Klinefelter nu prezintă semne până la pubertate , când caracteristicile fizice ale afecțiunii devin mai evidente; în unele cazuri nu există o simptomatologie evidentă, cu excepția sterilității sau, în orice caz, a unei reduceri puternice a fertilității , iar diagnosticul este formulat în consecință odată cu atingerea maturității sexuale.
La populația umană, starea 47, XXY este cea mai frecventă aneuploidie cromozomială sexuală la bărbați [5] . Cazurile de sindrom XXY pot apărea și la alte mamifere , cum ar fi șoarecii [6] . Aproximativ 80% dintre subiecții cu Klinefelter au cariotip 47, XXY, în timp ce restul de 20% din cazuri includ aneuploidii majore, mozaicuri 47, XXY / 46, XY și anomalii structurale ale cromozomului X.
Principalele manifestări includ hipogonadismul și scăderea fertilității . Alte diferențe fizice și comportamentale sunt, de asemenea, frecvente, deși severitatea lor variază de la individ la individ. În general, persoanele cu sindrom Klinefelter tind spre obezitate.
Note istorice și epidemiologie
Sindromul poartă numele lui Harry Klinefelter , care l-a descris pentru prima dată în 1942, lucrând cu Fuller Albright la Spitalul General Massachusetts din Boston [7] [8] . Înainte ca numele „sindrom Klinefelter” să intre în limbajul medical comun, afecțiunea a fost identificată ca aparținând „disgenezei tubulilor seminiferi ” [9] .
Sindromul, răspândit în mod egal în toate etniile , este cea mai frecventă tulburare genetică legată de heterozomi , cu o prevalență de 1-2 la 1000 de bărbați în populația generală [10] [11] [12] [13] . 3,1% dintre bărbații infertili sunt afectați [14] și, în plus, sindromul este principala cauză a hipogonadismului masculin [15] . Conform unei meta-analize, prevalența sindromului Klinefelter a crescut în ultimele decenii; cu toate acestea, această constatare nu pare a fi corelată cu creșterea vârstei medii a mamei la concepție, deoarece nu a fost observată nicio creștere a prevalenței celorlalte trisomii ale cromozomilor sexuali ( XXX și XYY ) [16] .
Etiologie


Un individ uman este caracterizat fiziologic prin doi cromozomi sexuali . Un bărbat cu cariotip normal are un cromozom X și un cromozom Y , acesta din urmă determinând sexul masculin; o femeie dimpotrivă doi cromozomi X. Factorul cauzal al sindromului Klinefelter este prezența a cel puțin unui cromozom X supranumerar [17] , care modifică nivelurile hormonale specifice ale bărbaților legate de fiziologia sexuală, acționând ca și cum ar fi al doilea cromozom X feminin, în special prin reducerea serului. valorile testosteronului . Prin urmare, un bărbat afectat de această patologie are o configurație genetică XXY care are atât perechea normală de cromozomi XY masculi, cât și cromozomii XX feminini.
Se crede că prezența unui cromozom X supranumerar este cauzată de un eveniment nedisjunct în timpul meiozei , care poate apărea fie pe partea maternă, fie pe cea paternă. Nu există factori de protecție care să împiedice acest lucru [18] .
În cazul în care cromozomul X suplimentar provine din partea paternă, evenimentul își are originea în timpul meiozei I ( gametogeneza ). Nedisjunctia apare atunci când cromozomii omologi, în acest caz X și Y, nu reușesc să se separe, așa cum ar trebui să se întâmple în spermatogeneză , producând un spermă cu doi cromozomi: unul X și unul Y. Fecundarea ulterioară a unui ou feminin normal (X) produce o descendență de tip XXY [19] . Aranjamentul cromozomului XXY este una dintre cele mai frecvente variații genetice ale cariotipului XY și apare la aproximativ 1 din 500 de bărbați născuți în viață [20] .
În caz contrar, dacă cromozomul supranumerar provine din partea maternă, nedisjunctia apare în timpul meiozei II . Acest lucru se întâmplă atunci când cromatidele surori dintr-un cromozom sexual feminin, în acest caz unul dintre cele două X-uri, nu reușesc să se separe. În consecință, se creează un ovul XX care, odată fertilizat cu un spermă Y, generează o descendență XXY. Se consideră că transmiterea maternă este mai frecventă decât transmiterea paternă [19] . Vârsta avansată a mamei este un factor predispozant, deși într-o măsură foarte limitată și semnificativ mai mică decât greutatea pe care o poate avea pentru sindromul Down [18] .
La mamiferele cu mai mult de un cromozom X, genele unuia dintre cei doi cromozomi X nu sunt exprimate : acest fenomen este cunoscut sub numele de inactivare a cromozomului X. Acest lucru se întâmplă la bărbații XXY, precum și la femelele XX normale [21] . Cu toate acestea, la bărbații XXY, unele gene localizate în regiunile pseudoautozomale ale cromozomilor X au gene omoloage corespunzătoare pe cromozomul Y și, prin urmare, pot fi exprimate [22] .
Prima descriere de caz a unui bărbat cu cariotip 47, XXY, publicată în 1959, a fost de Patricia Jacobs și John Strong de la Western General Hospital din Edinburgh , Scoția . Acest cariotip a fost găsit la un bărbat de 24 de ani care prezenta semne tipice ale sindromului Klinefelter [23] [24] .
Variante
Toate formele de sindrom Klinefelter se caracterizează prin prezența a cel puțin unui cromozom X supranumerar într-un fenotip masculin. Cu toate acestea, există unele variante [25] .
Cariotipul 48, XXYY apare în 1 caz la fiecare 18.000-40.000 nașteri masculine. Acest fenotip nu diferă mult de cel mai comun (47, XXY), cu excepția înălțimii medii mai mari găsite la vârsta adultă. Varianta 49, XXXXY este o polisomie rară care apare în aproximativ un caz pentru fiecare 85.000 de copii de sex masculin născuți. Această afecțiune este, în general, recunoscută la o vârstă fragedă din cauza deficitelor severe pe care le implică, incluzând: întârziere mentală marcată, dismorfism facial (gât scurt, ochi deschiși, gură și nas largi, strabism și urechi mari), criptorhidism , organe genitale ambigue și defecte scheletice ( cifoză , scolioză , coxa valga ) și cardiace [26] . Primul pacient afectat de această variantă a fost descris în 1960 [27] și de atunci până în 2012 au fost raportate puțin peste 100 de cazuri în literatură [26] .
În literatura de specialitate au fost raportate cazuri mult mai rare de cariotipuri 48, XXXY și 48, XXYY, caracterizate prin imagini fenotipice mai puțin severe comparativ cu 49, XXXXY, dar mai severe decât cariotipul clasic: apar malformații fizice, deficite de învățare și tulburări psihologice [28 ] [29] . De asemenea, au fost documentate cazuri foarte rare de variante, inclusiv cariotipuri: 49, XXXYY, 48, XYYY, 49, XYYYY și 49, XXYYY. Toate acestea implică aspect dismorf și întârziere mintală severă [25] [30] .
Bărbații cu sindrom Klinefelter pot avea mozaicism în cariotipul formei 47, XXY / 46, XY care implică grade diferite de spermatogeneză insuficientă [31] . Mosaicismul 47, XXY / 46, XX adaugă la fenotip celelalte trăsături clinice ale sindromului, dar se găsește foarte rar și, începând cu 2006, au fost descrise doar 10 cazuri [32] .
Clinica


Spectrul manifestărilor clinice este foarte larg în funcție de varianta sindromului: boala se poate prezenta cu o structură testiculară fetală, deficit de androgen, testicule și penis mic [33] [34] [35] [36] . Azoospermia este aproape întotdeauna prezentă la vârsta adultă [37] . Testiculele tind să fie mici la toate vârstele [38] [39] [40] datorită atât hipoplaziei celulelor germinale, cât și a celulelor interstițiale . În cele din urmă, ginecomastia este frecventă la persoanele cu sindrom Klinefelter [41] .
Spre deosebire de alte sindroame ale polisomiei cromozomiale X, care prezintă retard mental cu o prevalență mai mare, doar 10% dintre subiecții din sindromul Klinefelter prezintă retard mental [42] . Problemele cognitive sunt mai puțin răspândite și mai selective. Neurologic, sindromul Klinefelter este asociat cu dezvoltarea limbajului redusă, cu probleme de expresivitate, anomie , disartrie [43] [44] [45] [46] [47] . La nivel comportamental, se poate găsi imaturitatea, lipsa de încredere, timiditatea [48] [49] .
Genetica
La fel ca la indivizii feminini, cromozomul supranumerar X este inactivat aleatoriu, într-un fenomen numit lionizare . Este posibil ca inactivarea într-o celulă stem să fie apoi moștenită din populația de celule fiice, dar în general individul este un mozaic care poate avea, în principiu, jumătate din celule cu inactivarea unui cromozom X și jumătate cu inactivarea celălalt cromozom X. În acest sens, toate studiile care vizează evaluarea corelației simptomelor cu o anumită origine parentală a cromozomului X sunt inconsistente[50] .
Gena responsabilă de acest proces este XIST ( transcript specific X inactiv ), care transcrie un ARN exprimat numai de cromozomul inactivat și care nu codifică nicio proteină. Acționează asupra centrului de inactivare X (Xic, centrul de inactivare X ) și nu este exprimat la masculul cu cariotip normal (46, XY). A priori, fiecare dintre cei doi cromozomi X ai masculilor cu sindrom Klinefelter au aceeași probabilitate de a fi activat, dar nu toate genele sunt reduse la tăcere: 15% rămân în copii bialelelice duble [51] [38] și acestea par a fi primele responsabile fenotipul sindromului.
Cromozomul X diferă de autozomi în unele caracteristici legate de gene , care sunt scurte, mai puține ca număr și mai puțin prezente. Acestea sunt, de asemenea, caracterizate printr-un grad ridicat de conservare, dar nu de ordinul lor. Modelul polisomal , adică numărul de cromozomi X prezenți în cariotipul individului (45, X0, 47, XXY, 47, XXX, 48, XXXX și așa mai departe), se corelează puternic cu simptomele asociate [52] . Acest lucru a dus la ipoteza [53] unui rol activ al cromozomului X nu numai în determinarea sexului, ci și în dezvoltarea neurologică și în funcțiile creierului și comportamentale. Aceste date au fost confirmate de numeroase studii [54] [55] [56] [57] [58] [59] [60] [61] [62] . Genele cromozomului X sunt de fapt exprimate nu numai în primele etape ale spermatogenezei , ci și în mușchii scheletici , ovarele , placenta și creierul [63] [64] .
Gena RA este localizată în locusul Xq11-12, care codifică receptorul de androgen, un receptor nuclear cu un domeniu care recunoaște selectiv androgeni . Acest domeniu este codificat de o trăsătură extrem de polimorfă a primului exon, în care există o secvență bogată în triplete CAG. Proteinele cu o expansiune CAG scurtă sunt foarte asemănătoare și sensibile la androgeni [65] , dimpotrivă, o expansiune CAG lungă nu este foarte asemănătoare și, dacă repetarea depășește 40, apare o boală gravă, atrofia spinobulbară a lui Kennedy , cu afectare neuromusculară, marcată primar hipogonadism și ginecomastie de diferite grade. La bărbații cu sindrom Klinefelter, gena RA cu cea mai scurtă expansiune CAG [66] , adică cea mai activă RA, este inactivată. Prin urmare, subiecții nu numai că au o producție mai mică de androgeni, dar au și un receptor mai puțin similar.
Lungimea expansiunii CAG este până în prezent singurul parametru detectabil genetic care se corelează direct cu gradul mare de variabilitate fenotipică a sindromului Klinefelter [67] .
Dezvoltarea neurologică și comportamentală
Inteligența generală este măsurată de FSIQ ( IQ la scară completă ), care la bărbații Klinefelter se află în limite normale. Dacă, de fapt, coeficientul de inteligență la copii și adolescenți cu Klinefelter tinde să fie mai mic decât în grupurile de referință [68] [69] [70] [71] , la vârsta adultă poate să nu existe disparități. În schimb, există adesea o discrepanță între IQ verbal și IQ de performanță. Această discrepanță a fost observată atât la copii, cât și la adulții afectați [44] [69] [70] [71] [72] [73] .
Studiile sugerează că cel mai frecvent defect la băieții Klinefelter este verbal și apare evident în perioada școlară. La vârsta de 7 ani, individul are probleme moderate până la severe cu citirea, articularea cuvintelor, scrierea, în timp ce problemele de matematică apar puțin mai târziu [74] . Aproximativ 50-75% dintre copiii cu Klinefelter demonstrează unele dificultăți de învățare [75] și dintre acești 60-86% necesită educație direcționată sau asistată. [47] [69]
Adolescenții afectați prezintă încredere în sine redusă, sunt rezervați, au dificultăți în conținerea impulsurilor și acceptă reguli [36] . Aceste simptome au implicații pe termen lung pentru sociabilitate și activități școlare și pot persista până la maturitate. Cu toate acestea, dezadaptarea nu înseamnă o dezadaptare. Angajamentul în școală îl ajută adesea pe tânăr să depășească aceste probleme comportamentale [76] .
Alte manifestări clinice și fizice

Alte caracteristici fizice comune sunt circumferința redusă a craniului , părul corpului scurt, umerii îngustați și șoldurile largi, mușchiul slab [36] [41] , picioarele și brațele mai lungi decât în mod normal (astfel încât mâinile tale, cu brațele relaxate, să vină aproape în genunchi) ), voce puternică. Subiecții au un raport între degetul arătător și degetul inelar (numit și " raport cifră 2D: 4D") mai mare decât populația masculină și mai asemănător cu cel feminin (atât de mult încât subiecții, în comparație cu părinții lor respectivi , se constată adesea că au o statură mai mare, dar și degete mai scurte); Acest lucru se datorează nivelurilor scăzute de testosteron prenatal care afectează raportul de mai sus [77] . Simptome suplimentare includ riscul crescut de osteoporoză , tulburări autoimune ale tiroidei și diabet zaharat de tip 2 [78] . Există un risc crescut de 69% de a fi spitalizat chiar înainte de diagnosticarea Klinefelter [79] . Unele studii au asociat sindromul cu o înălțime ușor mai mare decât media și o predispoziție la supraponderalitate [41] [80] . Subiecții afectați au, de asemenea, o predispoziție la tromboza venoasă profundă , cu o prevalență de 8,6% la 50 de ani și 20,8% la 70 de ani [81] .
Pacienții cu sindrom Klinefelter suferă modificări hormonale deosebite [82] . Valorile serice ale hormonului foliculostimulant, hormonului luteinizant, hormonului anti-Müllerian și inhibinei B sunt normale în epoca prepubertală, în timp ce devin anormale în timp. Un studiu efectuat pe indivizi adulți cu sindrom a constatat niveluri scăzute de testosteron în 45% din cazuri și 43,6% dintre pacienți s-au plâns de disfuncție sexuală [80] .
Probabilitatea de a dezvolta tumori este deosebit, deoarece unele boli sunt strâns legate, cum ar fi primar mediastinale tumori ale celulelor germinale [83] și a cancerului de sân [84] , în timp ce pentru alții sindrom pare a fi un factor de protecție, cum ar fi în cazul lui cancer de prostată [85] . Utilizarea parametrului statistic ca „ risc absolut excesiv (EAR, risc exces absolut) la 100.000 de persoane pe an, este creșterea mortalității prin cancer pulmonar (AER +23.7), limfoame non-Hodgkin (AER +12, 1) și cancer de sân masculin ( AER +9,3) [85] .
Trim 48, XXXY este asociat cu gât scurt, epicantial falduri, clinodactyly , sinostoză radioulnara , și moderată la retard mental sever. Asociat cu cariotipul 49, XXXXY există o incidență crescută a anomaliilor cardiace congenitale , în special permeabilitatea botallo ductus arteriosus . Întârzierea mintală este adesea severă. Anomaliile scheletice includ sinostoză radioulnară, genunchiul valgus, pectus excavatum și clinodactilia [86] .
Fertilitate
Cariotipul 47, XXY se găsește la aproximativ 3,1% dintre bărbații azoospermici . Majoritatea bărbaților afectați sunt azoospermici și efectuarea unei biopsii testiculare poate evidenția absența celulelor germinale, hipertrofia celulei Leydig și fibroza marcată a tubilor seminiferi [87] . Mai mult, trebuie amintit că terapia cu androgeni, utilizată în tratamentul sindromului Klinefelter, afectează negativ fertilitatea [88] .
Au fost descrise sarcini concepute natural cu parteneri cu sindromul Klinefelter [89] și acest lucru este mai frecvent în cazurile de mozaicism. În cazurile de spermatogeneză reziduală prezentă, tehnicile de fertilizare asistată le pot oferi posibilitatea de a avea copii proprii, dar întrucât tehnica de biopsie a extracției spermei (extracția testiculară a spermei sau TESE) este invazivă, este întotdeauna necesar să se evalueze mai întâi prezența unii indicatori care sugerează posibila prezență a unei spermatogeneze reziduale. Găsirea spermatogoniei și spermatozoizilor este mai frecventă la tineri, de exemplu, deoarece numărul gametilor scade rapid odată cu vârsta, dar administrarea inhibitorilor de aromatază și a gonadotropinei corionice umane , pentru a stimula producția endogenă testiculară de testosteron, crește numărul de spermatozoizi. Fracțiile de spermă astfel obținute arată prezența unor gameți anormali, neeupoizi, de la 7 la 20% (la bărbații 46, XY această proporție este mai mică de 1%) [87] .
Diagnostic

Diagnosticul sindromului Klinefelter poate apărea înainte de naștere, în timpul vieții individului sau niciodată.
Diagnosticul prenatal, până de curând, era limitat la aproximativ 10% din cazuri [91] , dar răspândirea amniocentezei crește statisticile. Un sfert dintre bărbații afectați sunt recunoscuți în timpul pubertății [10] [37] , în timp ce 25% din cazuri sunt diagnosticați târziu la vârsta adultă [79] . În cele din urmă, se estimează [92] că aproximativ 64% dintre persoanele afectate de sindrom nu sunt recunoscute ca atare de-a lungul vieții.
Dacă diagnosticul prenatal apare după amniocenteză, adesea diagnosticul la bărbații adulți apare doar accidental, în contextul investigațiilor care nu sunt strict legate de afecțiune [93] , dar de obicei legate de investigațiile privind infertilitatea cuplului.
Manifestările clinice inițiale pot apărea în copilăria timpurie sau, mai des, în timpul pubertății și includ în general eșecul dezvoltării caracteristicilor sexuale secundare , microchidia și aspermatogeneza [7] , dar imaginea poate fi foarte variabilă, până la o mimică completă cu -Populația Klinefelter. Tendința către statura înaltă este dificil de diagnosticat în timpul pubertății [35] și, în ciuda prezenței testiculelor mici, analiza cariogramelor limfocitelor este standardul genetic pentru punerea diagnosticului [94] . În trecut, observarea corpului lui Barr era, de asemenea, o practică obișnuită [95] . Analiza cariotipului se efectuează și pe fibroblaste cutanate sau țesut testicular pentru a confirma mozaicismul [96] .
Alte metode de diagnostic includ detectarea nivelurilor serice crescute de gonadotropine (hormoni FSH și LH ), prezența azoospermiei și determinarea cromatinei sexuale în tampoane orale [97] . Un studiu din 1994 a propus, ca alternativă, utilizarea reacției în lanț a polimerazei (PCR) ca metodă de diagnostic mai rapidă. Testul este pozitiv dacă relevă prezența ARN-ului care poartă informațiile unei gene, conținute în cromozomul X, care servește ca un marker pentru inactivarea celui de-al doilea X (și al oricărui alt) suplimentar. Această genă, numită transcripție specifică X-inactivă (XIST), este transcrisă numai pe cromozomul X inactiv [97] .
Diagnostic diferentiat
Sindromul Klinefelter intră în diagnostic diferențial cu alte două afecțiuni genetice: sindromul X fragil (cauzat de o mutație a genei FMR1 pe cromozomul X) și sindromul Marfan (o tulburare autosomală dominantă care afectează țesutul conjunctiv ). Cauza hipogonadismului, tipică sindromului, poate fi atribuită multor alte afecțiuni medicale diferite [98] .
Au fost documentate cazuri rare de persoane cu sindrom Down care au prezentat și defectul 47 / XXY [99] .
Tratament
Mutația genetică este ireversibilă, totuși, pentru persoanele afectate care doresc să aibă un aspect mai masculin, este posibil să se recurgă la administrarea de testosteron [100] . Un studiu, efectuat pe pacienți adolescenți tratați cu implanturi subcutanate cu eliberare controlată a acestui hormon, a arătat rezultate bune în ciuda necesității unei monitorizări constante [101] . Terapia hormonală este, de asemenea, utilă pentru prevenirea apariției osteoporozei.
Bărbații cu ginecomastie și / sau hipogonadism suferă adesea de depresie și / sau anxietate socială [102] . Cel puțin un studiu recomandă oportunitatea sprijinului psihologic pentru tinerii cu sindrom Klinefelter pentru a reduce deficitul lor psihosocial [102] . Operația cu mastectomie poate fi luată în considerare atât pentru problemele psihologice datorate ginecomastiei, cât și pentru a reduce probabilitatea de a dezvolta cancer de sân [103] .
Utilizarea terapiei comportamentale poate atenua orice tulburări de limbaj, dificultăți școlare și de socializare. O abordare a terapiei ocupaționale este utilă la copiii cu sindrom care prezintă dispraxie motorie [104] .
Tratamentul infertilității

Până în 1996, bărbații cu un cariotip tipic sindromului Klinefelter erau considerați în general sterili . Cu toate acestea, începând cu 2010, s-au documentat peste 100 de sarcini cu FIV cu succes, cu spermatozoizi recoltați chirurgical de la bărbați cu sindrom ( extracția testiculară a spermei sau TESE) [105] .
Un studiu efectuat asupra rezultatelor 54 TESE a arătat că rata de recuperare a spermei este de 72% pentru fiecare procedură și că 69% dintre bărbați au avut un număr adecvat de spermatozoizi pentru a efectua injecția intracitoplasmatică de spermă . 46% din sarcinile obținute în acest mod s-au încheiat pozitiv și toți copiii născuți aveau un cariotip normal [106] . Aproximativ 30% dintre pacienții care suferă această procedură obțin aceste rezultate [107] .
În plus, crioconservarea materialului seminal prelevat în adolescență se realizează uneori pentru a încerca o posibilă procreare viitoare [108] .
Prognoză
Copiii cu forma XXY diferă puțin de copiii sănătoși. Deși este posibil să aibă de-a face cu probleme adesea emoționale și comportamentale în timpul adolescenței și să aibă dificultăți în realizarea școlii, cei mai mulți dintre ei pot obține independență deplină față de familiile lor la vârsta adultă. Unii reușesc să obțină o educație universitară și o viață aproape normală [109] .
Rezultatele unui studiu efectuat pe 87 de adulți australieni cu sindrom Klinefelter arată că cei care au avut un diagnostic și un tratament adecvat de la o vârstă fragedă au beneficiat semnificativ în comparație cu cei care au primit diagnosticul la vârsta adultă [110] .
Nu pare să existe o scădere substanțială a speranței de viață la persoanele cu sindrom Klinefelter. Au fost efectuate mai multe studii și au condus la rezultate nedefinitive. O primă lucrare publicată în 1985 a identificat o mortalitate mai mare datorată în principal bolii valvulare aortice , dezvoltării tumorilor și posibilelor hemoragii subarahnoidiene , precum reducerea speranței de viață cu aproximativ 5 ani [78] . Studiile ulterioare au redus această estimare prin asocierea stării cu o reducere mediană a supraviețuirii de 2,1 ani [111] . Cu toate acestea, aceste date nu sunt absolute și necesită verificări suplimentare [112] .
În urma diagnosticului sindromului, este necesară urmărirea [109] de către un endocrinolog.
Notă
- ^ Cotran, Ramzi S.; Kumar, Vinay; Fausto, Nelson; Nelso Fausto; Robbins, Stanley L.; Abbas, Abul K., Robbins și COTRAN bază patologic al bolii, St Louis, Mo, Elsevier Saunders, 2005, p. 179, ISBN 0-7216-0187-1 . .
- ^ Robert Bock, Înțelegerea sindromului Klinefelter: un ghid pentru bărbații XXY și familiile lor , pe NIH Pub. 93-3202 , Office of Research Reporting, NICHD, august 1993. Accesat la 7 aprilie 2007 (arhivat din original la 7 iulie 2007) .
- ^ Fundația Focus. Variații X și Y. thefocusfoundation.org Arhivat 13 ianuarie 2013 în Archive.is .
- ^ Sindromul Klinefelter . Informații despre sănătate , Institutul Național de Sănătate și Dezvoltare Umană, 19 februarie 2007. Accesat la 24 martie 2007 . și sindromul Klinefelter [ link broken ] , în Genetics Home Reference , Biblioteca Națională de Medicină, 2006. Accesat la 24 martie 2007 . ambele furnizează estimări statistice.
- ^ James, William; Berger, Timothy; Elston, Dirk, Boli ale pielii Andrews: Dermatologie clinică , 2005, p. 549, ISBN 0-7216-2921-0 . .
- ^ Liane Brauch Russell, Genetics of Mammalian Sex Chromosomes: mouse studies throw light on the functions and on the occasionally aberrant behavior of sex chromosomes , in Science , vol. 133, n. 3467, 9 giugno 1961, pp. 1795–1803, DOI : 10.1126/science.133.3467.1795 .
- ^ a b Klinefelter HF Jr, Reifenstein EC Jr, Albright F., Syndrome characterized by gynecomastia, aspermatogenesis without a-Leydigism and increased excretion of follicle-stimulating hormone , in J Clin Endocrinol Metab , vol. 2, n. 11, 1942, pp. 615–624, DOI : 10.1210/jcem-2-11-615 .
- ^ Klinefelter HF, Klinefelter syndrome: historical background and development , in South Med J , vol. 79, n. 45, 1986, pp. 1089–1093, DOI : 10.1097/00007611-198609000-00012 , PMID 3529433 .
- ^ ( EN ) Holub D, Grumbach M, Jailer J, Seminiferous tubule dysgenesis (Klinefelter's syndrome) in identical twins , in J Clin Endocrinol Metab , vol. 18, n. 12, dicembre 1958, pp. 1359-68, PMID 13611020 .
- ^ a b ( EN ) Bojesen A, Juul S, Gravholt CH, Prenatal and postnatal prevalence of Klinefelter syndrome: a national registry study , in Clin Endocrinol Metab , vol. 88, n. 2, 2003, pp. 622-6, PMID 12574191 .
- ^ ( EN ) Jacobs PA, Recurrence risks for chromosome abnormalities , in Birth Defects Orig Artic Ser , vol. 15, 5C, 1979, pp. 71-8, PMID 526617 .
- ^ ( EN ) MacLean et al, Abnormalities of sex chromosome constitution in newborn babies , in Lancet , vol. 19, 2(7199), agosto 1961, pp. 406-8, PMID 13764957 .
- ^ ( EN ) Visootsak J, Aylstock M, Graham JM Jr, Klinefelter syndrome and its variants: an update and review for the primary pediatrician , in Clin Pediatr (Phila) , vol. 40, n. 12, dicembre 2001, pp. 639-51, PMID 11771918 .
- ^ Mazzocco & Ross , p. 50 .
- ^ Matlach J, Grehn F, Klink T, Klinefelter Syndrome Associated With Goniodysgenesis , in J Glaucoma , gennaio 2012, DOI : 10.1097/IJG.0b013e31824477ef , PMID 22274665 .
- ^ Joan K Morris, Eva Alberman, Claire Scott e Patricia Jacobs, Is the prevalence of Klinefelter syndrome increasing? , in European Journal of Human Genetics , vol. 16, n. 2, 2007, pp. 163–170, DOI : 10.1038/sj.ejhg.5201956 .
- ^ De Leo, Fasano, Ginelli , p. 585 .
- ^ a b Cassidy-Allanson .
- ^ a b De Leo, Fasano, Ginelli , p. 586 .
- ^ Verri A, Cremante A, Clerici F, Destefani V, Radicioni A, Klinefelter's syndrome and psychoneurologic function , in Mol. Hum. Reprod. , vol. 16, n. 6, giugno 2010, pp. 425–33, DOI : 10.1093/molehr/gaq018 , PMID 20197378 .
- ^ Chow JC, Yen Z, Ziesche SM, Brown CJ, Silencing of the mammalian X chromosome , in Annu Rev Genomics Hum Genet , vol. 6, 2005, pp. 69–92, DOI : 10.1146/annurev.genom.6.080604.162350 , PMID 16124854 .
- ^ Blaschke RJ, Rappold G, The pseudoautosomal regions, SHOX and disease , in Curr. Opin. Genet. Dev. , vol. 16, n. 3, giugno 2006, pp. 233–9, DOI : 10.1016/j.gde.2006.04.004 , PMID 16650979 .
- ^ Jacobs PA, Strong JA, A case of human intersexuality having a possible XXY sex-determining mechanism , in Nature , vol. 183, n. 4657, 31 gennaio 1959, pp. 302–3, DOI : 10.1038/183302a0 , PMID 13632697 .
- ^ Jacobs PA,The William Allan Memorial Award address: human population cytogenetics: the first twenty-five annos , in Am J Hum Genet , vol. 34, n. 5, settembre 1982, pp. 689–98, PMC 1685430 , PMID 6751075 .
- ^ a b ( EN ) Linden MG, Bender BG, Robinson A, Sex chromosome tetrasomy and pentasomy , in Pediatrics , vol. 96, 4 Pt 1, ottobre 1995, pp. 672–82, PMID 7567329 .
- ^ a b ( EN ) Patacchiola F, Sciarra A, Di Fonso A, D'Alfonso A, Carta G, 49, XXXXY syndrome: an Italian child , in J. Pediatr. Endocrinol. Metab. , vol. 25, n. 1-2, 2012, pp. 165–6, PMID 22570969 .
- ^ ( EN ) Fraccaro M, Kaijser K, Lindsten J, A child with 49 chromosomes , in Lancet , vol. 2, n. 7156, ottobre 1960, pp. 899–902, PMID 13701146 .
- ^ ( EN ) Tartaglia N, Ayari N, Howell S, D'Epagnier C, Zeitler P,48,XXYY, 48,XXXY and 49,XXXXY syndromes: not just variants of Klinefelter syndrome , in Acta Paediatr. , vol. 100, n. 6, giugno 2011, pp. 851–60, DOI : 10.1111/j.1651-2227.2011.02235.x , PMC 3314712 , PMID 21342258 .
- ^ ( EN ) Frühmesser A, Kotzot D, Chromosomal variants in klinefelter syndrome , in Sex Dev , vol. 5, n. 3, 2011, pp. 109–23, DOI : 10.1159/000327324 , PMID 21540567 .
- ^ Nelson , pp. 1350-1355 .
- ^ ( EN ) Lim AS, Fong Y, Yu SL, Estimates of sperm sex chromosome disomy and diploidy rates in a 47,XXY/46,XY mosaic Klinefelter patient , in Hum. Genet. , vol. 104, n. 5, maggio 1999, pp. 405–9, PMID 10394932 .
- ^ ( EN ) Velissariou V, Christopoulou S, Karadimas C, Pihos I, Kanaka-Gantenbein C, Kapranos N, Kallipolitis G, Hatzaki A, Rare XXY/XX mosaicism in a phenotypic male with Klinefelter syndrome: case report , in Eur J Med Genet , vol. 49, n. 4, 2006, pp. 331–7, DOI : 10.1016/j.ejmg.2005.09.001 , PMID 16829354 .
- ^ ( EN ) Laron Z, Hochman IH, Small testes in prepubetal boys with Klinefelter's syndrome , in J Clin Endocrinol Metab , vol. 32, n. 5, maggio 1971, pp. 671-2, PMID 5577887 .
- ^ ( EN ) Zeger MP, Zinn AR, Lahlou N, Ramos P, Kowal K, Samango-Sprouse C, Ross JL, Effect of ascertainment and genetic features on the phenotype of Klinefelter syndrome , in J Pediatr , vol. 152, n. 5, maggio 2008, pp. 716-22, PMID 18410780 .
- ^ a b ( EN ) Salbenblatt JA, Bender BG, Puck MH, Robinson A, Faiman C, Winter JS, Pituitary-gonadal function in Klinefelter syndrome before and during puberty , in Pediatr Res , vol. 19, n. 1, gennaio 1985, pp. 82-6, PMID 3918293 .
- ^ a b c ( EN ) Stewart DA, Bailey JD, Netley CT, Park E, Growth, development, and behavioral outcome from mid-adolescence to adulthood in subjects with chromosome aneuploidy: the Toronto Study , in Birth Defects Orig Artic Ser , vol. 26, n. 4, 1990, pp. 131-88, PMID 2090316 .
- ^ a b ( EN ) Kamischke A, Baumgardt A, Horst J, Nieschlag E, Clinical and diagnostic features of patients with suspected Klinefelter syndrome , in J Androl , vol. 24, n. 1, Jan-Feb 2003, pp. 41-8, PMID 12514081 .
- ^ a b ( EN ) Wikström AM, Raivio T, Hadziselimovic F, Wikström S, Tuuri T, Dunkel L, Klinefelter syndrome in adolescence: onset of puberty is associated with accelerated germ cell depletion , in J Clin Endocrinol Metab , vol. 89, n. 5, maggio 2004, pp. 2263-70, PMID 15126551 .
- ^ ( EN ) Müller J, Skakkebaek NE, Ratcliffe SG, Quantified testicular histology in boys with sex chromosome abnormalities , in Int J Androl , vol. 18, n. 2, aprile 1995, pp. 57-62, PMID 7665210 .
- ^ ( EN ) Ratcliffe SG, The sexual development of boys with the chromosome constitution 47,XXY (Klinefelter's syndrome) , in Clin Endocrinol Metab , vol. 11, n. 3, novembre 1982, pp. 703-16, PMID 7139994 .
- ^ a b c Mazzocco & Ross , p. 52 .
- ^ Incidenza sindromi genetiche causa di ritardo mentale , su disabilitaintellettive.it . URL consultato il 21 ottobre 2012 .
- ^ ( EN ) Bender BG, Linden MG, Robinson A, Neuropsychological impairment in 42 adolescents with sex chromosome abnormalities , in Am J Med Genet , vol. 48, n. 3, ottobre 1993, pp. 169-73, PMID 8291574 .
- ^ a b ( EN ) Graham JM Jr, Bashir AS, Stark RE, Silbert A, Walzer S, Oral and written language abilities of XXY boys: implications for anticipatory guidance , in Pediatrics , vol. 81, n. 6, giugno 1988, pp. 795-806, PMID 3368277 .
- ^ ( EN ) Netley C, Rovet J, Hemispheric lateralization in 47,XXY Klinefelter's syndrome boys , in Brain Cogn , vol. 3, n. 1, gennaio 1984, pp. 10-8, PMID 6537238 .
- ^ ( EN ) Nielsen J, Johnsen SG, Sørensen K, Follow-up 10 annos later of 34 Klinefelter males with karyotype 47,XXY and 16 hypogonadal males with karyotype 46,XY , in Psychol Med , vol. 10, n. 2, maggio 1980, pp. 345-52, PMID 7384334 .
- ^ a b ( EN ) Walzer S, X chromosome abnormalities and cognitive development: implications for understanding normal human development , in J Child Psychol Psychiatry , vol. 26, n. 2, marzo 1985, pp. 177-84, PMID 3884639 .
- ^ ( EN ) Mandoki MW, Sumner GS, Hoffman RP, Riconda DL, A review of Klinefelter's syndrome in children and adolescents , in J Am Acad Child Adolesc Psychiatry , vol. 30, n. 2, marzo 1991, pp. 167-72, PMID 2016217 .
- ^ ( EN ) Ratcliffe SG, Bancroft J, Axworthy D, McLaren W, Klinefelter's syndrome in adolescence , in Arch Dis Child , vol. 57, n. 1, gennaio 1982, pp. 6-12, PMID 7065696 .
- ^ F. Tüttelmann, J. Gromoll, Novel genetic aspects of Klinefelter's syndrome. , in Mol Hum Reprod , vol. 16, n. 6, giugno 2010, pp. 386-95, DOI : 10.1093/molehr/gaq019 , PMID 20228051 .
- ^ Aksglaede L, Wikström AM, Rajpert-De Meyts E, Dunkel L, Skakkebaek NE, Juul A, Natural history of seminiferous tubule degeneration in Klinefelter syndrome , in Hum. Reprod. Update , vol. 12, n. 1, 2006, pp. 39–48, DOI : 10.1093/humupd/dmi039 , PMID 16172111 .
- ^ Davies W, Isles AR, Burgoyne PS, Wilkinson LS, X-linked imprinting: effects on brain and behaviour , in Bioessays , vol. 28, n. 1, gennaio 2006, pp. 35–44, DOI : 10.1002/bies.20341 , PMID 16369947 .
- ^ Skuse DH, James RS, Bishop DV, Coppin B, Dalton P, Aamodt-Leeper G, Bacarese-Hamilton M, Creswell C, McGurk R, Jacobs PA, Evidence from Turner's syndrome of an imprinted X-linked locus affecting cognitive function , in Nature , vol. 387, n. 6634, giugno 1997, pp. 705–8, DOI : 10.1038/42706 , PMID 9192895 .
- ^ Reik W, Walter J, Genomic imprinting: parental influence on the genome , in Nat. Rev. Genet. , vol. 2, n. 1, gennaio 2001, pp. 21–32, DOI : 10.1038/35047554 , PMID 11253064 .
- ^ Kato Y, Sasaki H, Imprinting and looping: epigenetic marks control interactions between regulatory elements , in Bioessays , vol. 27, n. 1, gennaio 2005, pp. 1–4, DOI : 10.1002/bies.20171 , PMID 15612042 .
- ^ Delaval K, Feil R, Epigenetic regulation of mammalian genomic imprinting , in Curr. Opin. Genet. Dev. , vol. 14, n. 2, aprile 2004, pp. 188–95, DOI : 10.1016/j.gde.2004.01.005 , PMID 15196466 .
- ^ Verona RI, Mann MR, Bartolomei MS, Genomic imprinting: intricacies of epigenetic regulation in clusters , in Annu. Rev. Cell Dev. Biol. , vol. 19, 2003, pp. 237–59, DOI : 10.1146/annurev.cellbio.19.111401.092717 , PMID 14570570 .
- ^ Haig D, Genomic imprinting and kinship: how good is the evidence? , in Annu. Rev. Genet. , vol. 38, 2004, pp. 553–85, DOI : 10.1146/annurev.genet.37.110801.142741 , PMID 15568986 .
- ^ Weinhäusel A, Haas OA, Evaluation of the fragile X (FRAXA) syndrome with methylation-sensitive PCR , in Hum. Genet. , vol. 108, n. 6, giugno 2001, pp. 450–8, PMID 11499669 .
- ^ Davies W, Isles AR, Wilkinson LS, Imprinted gene expression in the brain , in Neurosci Biobehav Rev , vol. 29, n. 3, maggio 2005, pp. 421–30, DOI :10.1016/j.neubiorev.2004.11.007 , PMID 15820547 .
- ^ Davies W, Isles AR, Wilkinson LS, Imprinted genes and mental dysfunction , in Ann. Med. , vol. 33, n. 6, settembre 2001, pp. 428–36, PMID 11585104 .
- ^ Zechner U, Wilda M, Kehrer-Sawatzki H, Vogel W, Fundele R, Hameister H, A high density of X-linked genes for general cognitive ability: a run-away process shaping human evolution? , in Trends Genet. , vol. 17, n. 12, dicembre 2001, pp. 697–701, PMID 11718922 .
- ^ ( EN ) Vallender EJ, Pearson NM, Lahn BT, The X chromosome: not just her brother's keeper , in Nat. Genet. , vol. 37, n. 4, aprile 2005, pp. 343–5, DOI : 10.1038/ng0405-343 , PMID 15800647 .
- ^ ( EN ) Ross MT, Grafham DV, Coffey AJ, et al. ,The DNA sequence of the human X chromosome , in Nature , vol. 434, n. 7031, marzo 2005, pp. 325–37, DOI : 10.1038/nature03440 , PMC 2665286 , PMID 15772651 .
- ^ Zinn AR, Ramos P, Elder FF, Kowal K, Samango-Sprouse C, Ross JL, Androgen receptor CAGn repeat length influences phenotype of 47,XXY (Klinefelter) syndrome , in J. Clin. Endocrinol. Metab. , vol. 90, n. 9, settembre 2005, pp. 5041–6, DOI : 10.1210/jc.2005-0432 , PMID 15956082 .
- ^ Zitzmann M, Depenbusch M, Gromoll J, Nieschlag E, X-chromosome inactivation patterns and androgen receptor functionality influence phenotype and social characteristics as well as pharmacogenetics of testosterone therapy in Klinefelter patients , in J. Clin. Endocrinol. Metab. , vol. 89, n. 12, dicembre 2004, pp. 6208–17, DOI : 10.1210/jc.2004-1424 , PMID 15579779 .
- ^ JC. Giltay, MC. Maiburg, Klinefelter syndrome: clinical and molecular aspects. , in Expert Rev Mol Diagn , vol. 10, n. 6, settembre 2010, pp. 765-76, DOI : 10.1586/erm.10.63 , PMID 20843200 .
- ^ Ratcliffe S,Long-term outcome in children of sex chromosome abnormalities , in Arch. Dis. Child. , vol. 80, n. 2, febbraio 1999, pp. 192–5, PMC 1717826 , PMID 10325742 .
- ^ a b c Bender BG, Puck MH, Salbenblatt JA, Robinson A, Dyslexia in 47,XXY boys identified at birth , in Behav. Genet. , vol. 16, n. 3, maggio 1986, pp. 343–54, PMID 3753369 .
- ^ a b Rovet J, Netley C, Bailey J, Keenan M, Stewart D, Intelligence and achievement in children with extra X aneuploidy: a longitudinal perspective , in Am. J. Med. Genet. , vol. 60, n. 5, ottobre 1995, pp. 356–63, DOI : 10.1002/ajmg.1320600503 , PMID 8546146 .
- ^ a b Walzer S, Bashir AS, Silbert AR, Cognitive and behavioral factors in the learning disabilities of 47,XXY and 47,XYY boys , in Birth Defects Orig. Artic. Ser. , vol. 26, n. 4, 1990, pp. 45–58, PMID 2090328 .
- ^ Porter ME, Gardner HA, DeFeudis P, Endler NS, Verbal deficits in Klinefelter (XXY) adults living in the community , in Clin. Genet. , vol. 33, n. 4, aprile 1988, pp. 246–53, PMID 3359682 .
- ^ Ratcliffe SG,Turner's syndrome , in Arch. Dis. Child. , vol. 61, n. 9, settembre 1986, p. 928, PMC 1778025 , PMID 3767428 .
- ^ Stewart DA, Netley CT, Bailey JD, Haka-Ikse K, Platt J, Holland W, Cripps M, Growth and development of children with X and Y chromosome aneuploidy: a prospective study , in Birth Defects Orig. Artic. Ser. , vol. 15, n. 1, 1979, pp. 75–114, PMID 444647 .
- ^ Rovet J, Netley C, Keenan M, Bailey J, Stewart D, The psychoeducational profile of boys with Klinefelter syndrome , in J Learn Disabil , vol. 29, n. 2, marzo 1996, pp. 180–96, PMID 8820203 .
- ^ Grace RJ, Klinefelter's syndrome: a late diagnosis , in Lancet , vol. 364, n. 9430, 2004, p. 284, DOI : 10.1016/S0140-6736(04)16679-8 , PMID 15262107 .
- ^ JT Manning, LP Kilduff e R. Trivers, Digit ratio (2D:4D) in Klinefelter's syndrome , in Andrology , vol. 1, n. 1, 2013-01, pp. 94–99, DOI : 10.1111/j.2047-2927.2012.00013.x . URL consultato l'11 aprile 2021 .
- ^ a b Price WH, Clayton JF, Wilson J, Collyer S, De Mey R,Causes of death in X chromatin positive males (Klinefelter's syndrome) , in J Epidemiol Community Health , vol. 39, n. 4, dicembre 1985, pp. 330–6, PMC 1052467 , PMID 4086964 .
- ^ a b Bojesen A, Juul S, Birkebaek NH, Gravholt CH, Morbidity in Klinefelter syndrome: a Danish register study based on hospital discharge diagnoses , in J. Clin. Endocrinol. Metab. , vol. 91, n. 4, aprile 2006, pp. 1254–60, DOI : 10.1210/jc.2005-0697 , PMID 16394093 .
- ^ a b Pacenza N, Pasqualini T, Gottlieb S, Knoblovits P, Costanzo PR, Stewart Usher J, Rey RA, Martínez MP, Aszpis S,Clinical Presentation of Klinefelter's Syndrome: Differences According to Age , in Int J Endocrinol , vol. 2012, 2012, p. 324835, DOI : 10.1155/2012/324835 , PMC 3265068 , PMID 22291701 .
- ^ Bengt Zöller, Jianguang Ji e Jan Sundquist, High Risk of Venous Thromboembolism in Klinefelter Syndrome , in Journal of the American Heart Association: Cardiovascular and Cerebrovascular Disease , vol. 5, n. 5, 20 maggio 2016, DOI : 10.1161/JAHA.116.003567 . URL consultato il 22 marzo 2021 .
- ^ Tincani BJ, Mascagni BR, Pinto RD, Guaragna-Filho G, Castro CC, Sewaybricker LE, Viguetti-Campos NL, Marques-de-Faria AP, Maciel-Guerra AT, Guerra-Júnior G., Klinefelter syndrome: an unusual diagnosis in pediatric patients. , in J Pediatric , luglio 2012, PMID 22915094 .
- ^ Hainsworth JD, Greco FA, Germ cell neoplasms and other malignancies of the mediastinum , in Cancer Treat. Res. , vol. 105, 2001, pp. 303–25, PMID 11224992 .
- ^ Swerdlow AJ, Hermon C, Jacobs PA, Alberman E, Beral V, Daker M, Fordyce A, Youings S, Mortality and cancer incidence in persons with numerical sex chromosome abnormalities: a cohort study , in Ann. Hum. Genet. , vol. 65, Pt 2, marzo 2001, pp. 177–88, DOI : 10.1017/S0003480001008569 , PMID 11427177 .
- ^ a b Swerdlow AJ, Schoemaker MJ, Higgins CD, Wright AF, Jacobs PA, Cancer incidence and mortality in men with Klinefelter syndrome: a cohort study , in J. Natl. Cancer Inst. , vol. 97, n. 16, agosto 2005, pp. 1204–10, DOI : 10.1093/jnci/dji240 , PMID 16106025 .
- ^ Peet J,Weaver DD, Vance GH. 49, XXXXY: a distinct phenotype. Three new cases and revies. J Med Genet 1998; 35:420-424.
- ^ a b ( EN ) Ferlin A, Garolla A, Foresta C, Chromosome abnormalities in sperm of individuals with constitutional sex chromosomal abnormalities , in Cytogenet. Genome Res. , vol. 111, n. 3-4, 2005, pp. 310–6, DOI : 10.1159/000086905 , PMID 16192710 .
- ^ ( EN ) Lanfranco F, Kamischke A, Zitzmann M, Nieschlag E, Klinefelter's syndrome , in Lancet , vol. 364, n. 9430, 2004, pp. 273–83, DOI : 10.1016/S0140-6736(04)16678-6 , PMID 15262106 .
- ^ Forti G, Csilla K, La fertilità nella sindrome di Klinefelter: implicazioni pratiche e terapia , in L'Endocrinologo , vol. 7, 2006, pp. 32-9.
- ^ ( EN ) Bojesen A, Gravholt CH, Klinefelter syndrome in clinical practice , in Nat Clin Pract Urol , vol. 4, n. 4, aprile 2007, pp. 192–204, DOI : 10.1038/ncpuro0775 , PMID 17415352 .
- ^ ( EN ) Abramsky L, Chapple J, 47,XXY (Klinefelter syndrome) and 47,XYY: estimated rates of and indication for postnatal diagnosis with implications for prenatal counselling , in Prenat Diagn , vol. 17, n. 4, aprile 1997, pp. 363-8, PMID 9160389 .
- ^ ( EN ) Smyth CM, Bremner WJ, Klinefelter syndrome , in Arch Intern Med , vol. 158, n. 12, 22 giugno 1998, pp. 1309-14, PMID 9645824 .
- ^ ( PL ) Grzywa-Celińska A, Rymarz E, Mosiewicz J, [Diagnosis differential of Klinefelter's syndrome in a 24-anno old male hospitalized with with sudden dyspnoea--case report] , in Pol. Merkur. Lekarski , vol. 27, n. 160, ottobre 2009, pp. 331–3, PMID 19928664 .
- ^ PA. Jacobs, The incidence and etiology of sex chromosome abnormalities in man. , in Birth Defects Orig Artic Ser , vol. 15, n. 1, 1979, pp. 3-14, PMID 444646 .
- ^ Kamischke A, Baumgardt A, Horst J, Nieschlag E, Clinical and diagnostic features of patients with suspected Klinefelter syndrome , in J. Androl. , vol. 24, n. 1, 2003, pp. 41–8, PMID 12514081 .
- ^ ( CS ) Kurková S, Hána V, Mayerová K, Pacovská K, Musilová J, Stĕpán J, Michalová K e Zemanová Z, Molecular cytogenetic diagnosis of Klinefelter's syndrome in men more frequently detects sex chromosome mosaicism than classical cytogenetic methods , in Cas. Lek. Cesk. , vol. 138, n. 8, aprile 1999, pp. 235–8, PMID 10510542 .
- ^ a b ( EN ) Kleinheinz A, Schulze W, Klinefelter's syndrome: new and rapid diagnosis by PCR analysis of XIST gene expression , in Andrologia , vol. 26, n. 3, 1994, pp. 127–9, PMID 8085664 .
- ^ ( EN ) Harold Chen, Klinefelter Syndrome Differential Diagnoses , su emedicine.medscape.com , medscape.com. URL consultato il 18 agosto 2012 .
- ^ Sanz-Cortés M, Raga F, Cuesta A, Claramunt R, Bonilla-Musoles F, Prenatally detected double trisomy: Klinefelter and Down syndrome , in Prenat. Diagn. , vol. 26, n. 11, novembre 2006, pp. 1078–80, DOI : 10.1002/pd.1561 , PMID 16958145 .
- ^ Wikström AM, Dunkel L, Klinefelter syndrome , in Best Pract. Res. Clin. Endocrinol. Metab. , vol. 25, n. 2, aprile 2011, pp. 239–50, DOI : 10.1016/j.beem.2010.09.006 , PMID 21397196 .
- ^ ( EN ) Moskovic DJ, Freundlich RE, Yazdani P, Lipshultz LI, Khera M, Subcutaneous implantable testosterone pellets overcome noncompliance in adolescents with klinefelter syndrome , in J Androl , vol. 33, n. 4, luglio 2012, pp. 570-3, PMID 21940986 .
- ^ a b Simm PJ, Zacharin MR, The psychosocial impact of Klinefelter syndrome--a 10 anno review , in J. Pediatr. Endocrinol. Metab , vol. 19, n. 4, aprile 2006, pp. 499–505, PMID 16759035 .
- ^ Gabriele R, Borghese M, Conte M, Egidi F, [Clinical-therapeutic features of gynecomastia] , in G Chir , vol. 23, n. 6-7, 2002, pp. 250–2, PMID 12422780 .
- ^ ( EN ) Harold Chen, Klinefelter Syndrome - Treatment , su emedicine.medscape.com , medscape.com. URL consultato il 4 settembre 2012 .
- ^ Fullerton G, Hamilton M, Maheshwari A., Should non-mosaic Klinefelter syndrome men be labelled as infertile in 2009? , in Hum Reprod. , vol. 25, n. 3, 2010, pp. 588–97, DOI : 10.1093/humrep/dep431 , PMID 20085911 .
- ^ Schiff JD, Palermo GD, Veeck LL, Goldstein M, Rosenwaks Z, Schlegel PN, Success of testicular sperm extraction [corrected] and intracytoplasmic sperm injection in men with Klinefelter syndrome , in J. Clin. Endocrinol. Metab. , vol. 90, n. 11, novembre 2005, pp. 6263–7, DOI : 10.1210/jc.2004-2322 , PMID 16131585 .
- ^ ( EN ) Vignozzi L, Corona G, Forti G, Jannini EA, Maggi M, Clinical and therapeutic aspects of Klinefelter's syndrome: sexual function , in Mol. Hum. Reprod. , vol. 16, n. 6, giugno 2010, pp. 418–24, DOI : 10.1093/molehr/gaq022 , PMID 20348547 .
- ^ Damani MN, Mittal R, Oates RD, Testicular tissue extraction in a young male with 47,XXY Klinefelter's syndrome: potential strategy for preservation of fertility , in Fertil. Steril. , vol. 76, n. 5, novembre 2001, pp. 1054–6, PMID 11704135 .
- ^ a b ( EN ) Medscape - follow-up , su emedicine.medscape.com . URL consultato il 19 agosto 2012 .
- ^ ( EN ) Herlihy AS, McLachlan RI, Gillam L, Cock ML, Collins V, Halliday JL, The psychosocial impact of Klinefelter syndrome and factors influencing quality of life , in Genet. Med. , vol. 13, n. 7, luglio 2011, pp. 632–42, DOI : 10.1097/GIM.0b013e3182136d19 , PMID 21546843 .
- ^ ( EN ) Bojesen A, Juul S, Birkebaek N, Gravholt CH, Increased mortality in Klinefelter syndrome [ collegamento interrotto ] , in J. Clin. Endocrinol. Metab. , vol. 89, n. 8, agosto 2004, pp. 3830–4, DOI : 10.1210/jc.2004-0777 , PMID 15292313 .
- ^ ( EN ) Swerdlow AJ, Higgins CD, Schoemaker MJ, Wright AF, Jacobs PA,Mortality in patients with Klinefelter syndrome in Britain: a cohort study [ collegamento interrotto ] , in J. Clin. Endocrinol. Metab. , vol. 90, n. 12, dicembre 2005, pp. 6516–22, DOI : 10.1210/jc.2005-1077 , PMID 16204366 .
Bibliografia
- Giacomo De Leo, Silvia Fasano, Enrico Ginelli, Biologia e genetica , EdiSES, 2009, ISBN 978-88-7959-563-6 .
- Michel Godfryd. Klinefelter (sindrome di) , in Dizionario di psicologia e psichiatria . 1ª ed. Roma, Newton Compton editori (collana Il sapere - Enciclopedia tascabile Newton - Sezione di scienze umane - 18), 1994. p. 50. ISBN 88-7983-487-8 .
- ( EN ) Virginia Isaacs, Living with Klinefelter Syndrome, Trisomy X and 47,XYY: A Guide for Families and Individuals Affected by Extra X and Y Chromosomes , 2012, ISBN 978-0-615-57400-4 .
- ( EN ) Suzanne B. Cassidy, Judith E. Allanson, Management of Genetic Syndromes , Wiley-Liss, 2001, ISBN 978-0-471-31286-4 .
- ( EN ) Waldo Emerson Nelson, Nelson textbook of pediatrics , Saunders, 1975, ISBN 978-0-7216-9018-6 .
- ( EN ) Michele Mazzocco, Judith Ross, Neurogenetic Developmental Disorders: Variation of Manifestation in Childhood , MIT Press, 2007, ISBN 978-0-262-13480-4 .
- ( EN ) Michael Zitzmann, Lise Aksglaede e Giovanni Corona, European academy of andrology guidelines on Klinefelter Syndrome: Endorsing Organization: European Society of Endocrinology , in Andrology , 6 ottobre 2020, pp. andr.12909, DOI : 10.1111/andr.12909 . URL consultato il 24 ottobre 2020 .
Voci correlate
Altri progetti
-
 Wikimedia Commons contiene immagini o altri file su Sindrome di Klinefelter
Wikimedia Commons contiene immagini o altri file su Sindrome di Klinefelter
Collegamenti esterni
- Sindrome di Klinefelter , su sapere.it , De Agostini .
- ( EN ) Sindrome di Klinefelter , su Enciclopedia Britannica , Encyclopædia Britannica, Inc.
| Classificazione e risorse esterne ( EN ) | ICD-9-CM : 758.7 ; ICD-10-CM : Q98.4 e Q98.0 ; MeSH : D007713 ; MedlinePlus : 000382 ; eMedicine : 945649 ; |

| Controllo di autorità | LCCN ( EN ) sh85072641 · GND ( DE ) 4164211-9 |
|---|