Sindromul Marfan
| Sindromul Marfan | |
|---|---|
| Boala rara | |
| Cod. SSN | RN1320 |
| Specialitate | genetică clinică |
| Clasificare și resurse externe (EN) | |
| OMIM | 154700 |
| Plasă | D008382 |
| MedlinePlus | 000418 |
| eMedicină | 946315 și 1258926 |
| GeneReviews | Prezentare generală |
| Eponime | |
| Antoine Marfan | |
Sindromul Marfan este o boală autozomală dominantă (MIM / OMIM 154700) care afectează țesutul conjunctiv [1] . Sindromul este caracterizat printr - o producție anormală a fibrillin1 proteinei cauzata de o mutatie a FBN1 genei [2] localizat pe cromozomul 15 [3] [4] . Manifestările sindromului Marfan afectează organele care conțin țesut conjunctiv, cum ar fi sistemul osos , ochii , inima și vasele de sânge , plămânii și membranele fibroase care acoperă creierul și coloana vertebrală . Termenul provine de la numele pediatrului francez Antoine Marfan , care a descris prima dată sindromul în 1896 la o fetiță de 5 ani.
Epidemiologie
Se estimează că un copil din fiecare 5000 de nașteri vii este afectat de sindrom, dar incidența este probabil subestimată din cauza formelor frecvente de biciuire , care, ne manifestându-se cu un fenotip specific , pot scăpa cu ușurință de diagnostic. Masculii și femelele sunt la fel de afectate; nu există distincții de rasă în incidența bolii. Au fost indicate mai multe figuri istorice ca fiind probabil afectate: Abraham Lincoln , Charles de Gaulle , Niccolò Paganini , faraonul Akhenaton , Charles Maurice de Talleyrand-Périgord , Reinhard Heydrich .
Aproximativ 75% dintre afectați au cel puțin un părinte afectat la rândul său, cu transmitere genică autosomală dominantă; cu toate acestea, în 25% din cazuri, nu există alți pacienți cu Marfan în familia lor: în acest caz este deci o mutație genetică de novo în spermatozoizii paterni. Se pare că mutațiile de novo sunt mai frecvente dacă vârsta tatălui conceputului depășește 45 de ani. [5]
semne si simptome
Spectrul manifestărilor sindromului este foarte larg și diversificat: în timp ce pentru unii pacienți diagnosticul este imediat și precoce, pentru acei subiecți pentru care sunt prezente doar unele dintre simptome diagnosticul este dificil. Doar o investigație genetică (cu sensibilitate foarte mare) poate garanta în cele din urmă un diagnostic precis pentru aceste persoane care, paradoxal, sunt cele mai expuse riscului din cauza absenței unei monitorizări periodice atente a sistemului cardiovascular. [6]
Sistemul locomotor
În demonstrațiile musculare - scheletice , ceea ce este cel mai izbitor este înălțimea pacienților, care sunt de obicei mai mari decât media colegilor și adesea dezvoltă (mai ales în timpul adolescenței ) semne de emaciație excesivă (înțeleasă nu numai ca o simplă subponderalitate , ci și ca un habitus excesiv conic și înclinat în general: acest lucru se datorează în principal membrelor deosebit de lungi și subțiri în comparație cu trunchiul, aspect numit „ dolicostenomelia ”: acesta este așa-numitul habitus marfanoid. Un studiu realizat în Coreea de Sud arată că 50 percentila înălțimii adulților Marfan depășește percentila 97 a populației generale, în timp ce percentila 50 a greutății Marfan depășește ușor percentila 75 a populației sănătoase la bărbați și se află între percentila 50 și 75 a populației sănătoase la femei; practic greutate corporală redusă în raport cu o înălțime care este aproape întotdeauna peste normă. [7] musculatura este adesea redusă cu habitus ectomorf și dificultăți în dobândirea masei musculare, chiar dacă fără hipoplazie severă - mai gravă este de obicei deficiența musculară observată în sindromul Beals . Poate fi prezentă o rezistență redusă la efort fizic.
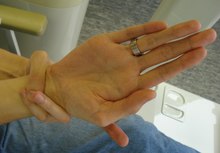
O altă caracteristică este lungimea, hiperextensibilitatea și forma conică a degetelor, care se numește „ arahnodactilie ” și este recunoscută prin semnul degetului mare sau semnul lui Steinberg (întreaga miniatură flexată spre palma mâinii trece de marginea ulnară ) și de încheietura mâinii semn sau semn Walker-Murdoch (vârful degetului mic și degetul mare al aceleiași mâini se pot suprapune făcând o întoarcere în jurul încheieturii mâinii celuilalt braț); arahnodactilia, care tinde să provoace hipermobilitatea degetelor, este legată de laxitatea generalizată a ligamentelor [6] . Frecvente în boli precum sindromul Marfan și boala osoasă Paget , dar foarte rară la pacienții altfel sănătoși, este proeminența acetabulului , care se agravează adesea odată cu vârsta. Această proeminență, împreună cu ectazia durală și, în general, cu predispoziție la dezvoltarea oboselii cronice, îi determină pe mulți care suferă de sindrom să dezvolte dureri cronice și lipsă de energie de-a lungul anilor; Există și degenerescență osoasă, cu osteopenie și, în unele cazuri, osteoporoză cu debut mai timpuriu decât populația generală.
Deformitățile toracice sunt foarte frecvente și deosebite, datorate nu atât unei deformări a sternului , ci mai degrabă unei supradezvoltări și unei consecințe flexii exterioare sau interioare a coastelor ; aceste anomalii se numesc respectiv piept cu cheile și pieptul excavat : sunt adesea foarte evidente, cu toate acestea numai în cele mai izbitoare cazuri împiedică atingerea capacității pulmonare maxime la inhalare (și, prin urmare, trebuie rezolvate cu o intervenție chirurgicală) . [8] Coexistența cifozei și a scoliozei peste 20 °, care tind să se înrăutățească odată cu vârsta, este, de asemenea, recurentă în sindrom; pentru o scolioză care depășește 40 ° -45 ° este deseori recomandată o intervenție chirurgicală corectivă. La pacienții Marfan este , de asemenea găsit la nivelul podologic , o incidență crescută a piciorului plat și deformarea hindfoot , cu toc flectat spre exterior; hallux valgus nu este, de asemenea, neobișnuit. O altă caracteristică ortopedică destul de frecventă în sindrom este genu recurvatum , adică genunchii mișcați înapoi față de planul vertical al membrului inferior.
La nivel dentar și gnatologic , sunt foarte frecvente micrognatia , retrognatia , palatul ogival (palatul îngust) cu malocluzie consecventă (adesea de tip 2) și hipoplazia maxilară . Toate acestea cresc probabilitatea de a avea un set de dinți anormal, cu dinți încrucișați, în special în arcul superior . Aceste anomalii orale, combinate cu slăbiciunea intrinsecă a mușchilor căilor respiratorii superioare , sunt responsabile de o incidență mai mare a apneei obstructive de somn la pacienții cu Marfan decât la populația generală. [9]
Sistemul cardiovascular
Aparatul L 'cardiovascular cuprinde uneori modificări foarte grave: porțiunea ascendentă a aortei tinde să se dilate, rezultând în slăbirea mediului tunic de sânge care, cu un cerc vicios care ține cu tot mai multe dificultăți tensiunea arterială, aducând expansiunea să crească progresiv, devenind anevrismal după o fază inițială de ectazie . Apariția unei disecții a aortei ascendente, mult mai frecventă în cazul unui anevrism complet, dar și posibilă cu diametre aortice în limitele normale, este tipică sindromului și tinde să apară până la vârsta mijlocie (în principal bulbul aortic și rădăcina aortică ). Dezvoltarea anevrismelor în arcada aortică sau în aorta descendentă , inclusiv aorta abdominală , este de asemenea mai puțin frecventă, dar nu mai puțin frecventă. În timp ce un pacient diagnosticat va fi urmărit pe tot parcursul evoluției bolii și apoi monitorizat cu o ecocardiogramă pentru orice modificare a măsurătorilor sale aortice (și, dacă este necesar, operat înainte de disecție), persoanele care nu primesc un diagnostic riscă să fie afectate brusc. de patologia aortică cu consecințe deseori dramatice. [6] 70% dintre cei afectați de sindromul Marfan ajung să prezinte un diametru aortic anormal în 20 de ani de viață, 80% în 40 de ani, 35% în primii 5 ani de viață. [10]
Diametrul rădăcinii aortice este direct proporțional cu riscul de disecție și ruptură aortică: aorta pacienților cu Marfan are o fragilitate intrinsecă și, prin urmare, este expusă riscului de disecție chiar și cu diametre în limitele normale, totuși riscul de evenimente aortice, în caz de terapie beta-blocantă adecvată și abținere de la efort fizic intens, acesta rămâne cu mult sub 1% pe an cu diametre ascendente ale aortei mai mici de 50 mm, în timp ce crește exponențial dincolo de acest prag, devenind aproximativ 5% pe an în cazul diametrului în jur de 60 mm. [11]
Să ne amintim de insuficiența valvei mitrale (prezentă la 50-60% dintre pacienții cu Marfan) și a tricuspidului ; uneori aceste valvulopatii se datorează prolapsului lambourilor, uneori ruperii cordoanelor tendinoase. [6] Valva aortică este, de asemenea, adesea insuficientă; sunt frecvente insuficiența mitrală, aortică și tricuspidă. Boala severă a valvei cardiace poate duce la insuficiență cardiacă congestivă . Prolapsul mitral progresează adesea odată cu vârsta, ducând la dehiscența valvei în cazurile severe. [12] Această anomalie valvulară poate fi corectată prin intervenție chirurgicală (care poate fi efectuată cu înlocuirea valvei biologice cu una mecanică sau cu menținerea valvelor biologice), după care rata de supraviețuire este excelentă și riscul aritmogen pe termen lung este scăzut. [13]
Prezența aritmiilor ventriculare majore (inclusiv tahicardie ventriculară susținută sau nesustenată , precum și frecvente bătăi ectopice ventriculare ), atât în absență, cât și în comorbiditate cu boală valvulară semnificativă, expune pacienților Marfan un risc mai mare de deces subit cardiac , crescând mortalitatea acestora pentru stop cardiac și pentru toate cauzele comparativ cu Marfan cu manifestări aritmice mai ușoare sau absente. [14] Pacienții cu Marfan prezintă un risc crescut de a dezvolta endocardită infecțioasă cu afectări în principal ale valvei mitrale; pentru a reduce această posibilitate, terapia cu antibiotice este recomandată în scop preventiv înainte de a fi supusă oricărui tip de intervenție chirurgicală, în special a celor ortodontice .
Ochi

Manifestările oculare se caracterizează fundamental prin deplasarea lentilei (numită și ectopia lentis ) în urma unei subluxații a acesteia și prin sferofacie [15] , adesea prezentă în formele diagnosticate în copilărie și de probleme retiniene , precum detașarea . [6] Dislocarea lentilei, în special, este un semn foarte particular al sindromului: poate apărea la orice vârstă și poate fi unilateral sau bilateral. Când apare la bătrânețe, tinde să fie mai severă, bilaterală și cu o evoluție deosebit de rapidă. Anomaliile oculare menționate anterior cauzează foarte adesea erori de refracție: printre ele cea mai frecventă este deseori miopia foarte puternică, adesea cu debut în copilărie , dar pot fi găsite și presbiopia și astigmatismul . [16] Miopia poate apărea în cele mai severe cazuri de subluxare a lentilelor. Strabismul , atât exotrop cât și exotrop, este de asemenea frecvent , în general la debutul copilăriei. [17] Colobomul iris este, de asemenea, mai frecvent în sindromul Marfan. Sindromul vă pune un risc destul de mare de a dezvolta cataractă cu mult înainte de bătrânețe și glaucom la orice vârstă.
Alte manifestări
Un alt semn tipic, aproape patognomonic , al sindromului este ectazia durală , care apare la mai mult de jumătate dintre pacienții cu Marfan pe parcursul vieții și care constă în dilatarea progresivă a căptușelii duramater în jurul coloanei vertebrale ; în general apare la nivel lombosacral, unde presiunea exercitată de lichidul cefalorahidian asupra țesuturilor care îl conțin este mai mare. În etapele inițiale, ectazia durală tinde să fie asimptomatică, dar atunci când se agravează tinde să provoace dureri lombosacrale , cefalee , astenie , slăbiciune și sensibilitate scăzută la nivelul membrelor inferioare, dureri rectale și genitale, incontinență urinară, retenție urinară ; simptomele tind să se înrăutățească atunci când stați în poziție verticală. Rezonanța magnetică nucleară sau tomografia computerizată sunt necesare pentru diagnosticarea acestei modificări.
La nivel pulmonar, pe lângă posibilele dificultăți de respirație cauzate de formele severe ale deformărilor toracice menționate mai sus, pneumotoraxul se găsește cu o incidență crescută, în special la subiecții tineri: evenimente multiple de pneumotorax spontan sunt posibile la pacienții cu Marfan, dar sunt foarte rare în alt mod. subiecți sănătoși. Chiar și emfizemul, deși fără factori de risc asociați, cum ar fi fumatul , este destul de frecvent în rândul celor afectați de sindrom, cu modificări ale auscultației, în special la nivelul vârfurilor pulmonare. Doar 37% dintre cei afectați au funcție pulmonară normală: 19% au un model restrictiv și 44% au chiar un model de obstrucție respiratorie. [18]
Dungile cutanate de pe membre se găsesc foarte des la nivelul membrelor care apar în preadolescență și pot să nu fie legate de modificări mari ale înălțimii sau greutății corporale: ele tind, de asemenea, să fie localizate în locații atipice, cum ar fi membrele superioare și umerii [ 19] ; inițial roșii, devin progresiv albi și mai puțin inestetici, dar aproape întotdeauna devin cronici.
Aproximativ 29% dintre persoanele cu sindrom au modificări semnificative ale coagulării : modificările structurii trombocitelor pot duce la sângerări excesive, similare cu cele observate în boala von Willebrand sau în alte trombocitopatii congenitale. [20] S-a demonstrat o proporționalitate directă între concentrația sanguină a dimerului D și rata naturală de progresie a dilatației aortice la pacienții adulți cu Marfan, precum și o proporționalitate inversă între gradul de activitate al factorului VIII de coagulare și rata de dilatație aortică. . Cu sindromul Marfan, factorul von Willebrand și același factor VIII de coagulare au în medie o activitate mai mică decât în populația generală, în timp ce timpul de tromboplastină parțială (PTT) nu este în general modificat. [21]
Laxitatea generală a țesutului conjunctiv favorizează formarea herniilor , în special a herniilor inghinale , care ar fi prezentă la aproximativ 32% dintre pacienții cu Marfan peste 26 de ani care sunt supuși unei intervenții chirurgicale pentru a înlocui aorta ascendentă. [22] Dezvoltarea herniei hiatale , a herniei ombilicale - chiar mai frecvente în sindromul Ehlers-Danlos de tip vascular și a bolii de reflux gastroesofagian (doar uneori legate de prezența unei hernii hiatale) sunt mai frecvente în populația Marfan decât în populație sănătoasă.
Sindromul neonatal Marfan
Aproximativ 5-10% dintre sindroamele Marfan se manifestă cu simptome grave și care se agravează rapid de la naștere: acesta este așa-numitul sindrom Marfan neonatal (care nu trebuie confundat cu sindromul clasic Marfan dacă acesta din urmă este diagnosticat în perioada neonatală). Cursul este mult mai sever decât în sindromul clasic Marfan și se observă de la naștere anomalii craniofaciale și oculare, inclusiv iridodoneză , megalocornea , ectopia lentis , auriculă mototolită - similar cu ceea ce se observă în sindromul Beals mult mai puțin periculos -, exces de piele la dați un aspect „senil” feței copilului afectat; se mai observă în contracturi multiple ale articulațiilor de nivel, agravarea emfizemului pulmonar și defecte valvulare frecvente foarte grave, inclusiv prolapsul valvei mitrale și valva tricuspidă ; aorta ascendentă tinde să se dilate încă din primele luni de viață și este supusă rupturilor anevrismelor din copilăria timpurie. Afectarea pulmonară este gravă, din cauza insuficienței cardiace severe: arterele pulmonare dilatate sunt observate într-o imagine a hipertensiunii pulmonare severe. De asemenea, se găsesc frecvent pectus carinatum , arahnodactilia și dolichostenomelia . [23] Similar sindromului clasic Marfan, varianta neonatală este consecința unei mutații a genei FBN1; [24] prognosticul formei neonatale a bolii este foarte sever: 95% dintre pacienți nu depășesc un an de viață, cu o mediană de aproximativ 5 luni; cu toate acestea, speranța medie de viață este de 16 luni, reflectând modul în care există unele valori aberante care pot supraviețui peste 4-11 ani, ajungând la adolescență sau chiar la vârsta adultă timpurie. [25]
Diagnostic
Criterii de diagnostic Ghent
În 2010, nosologia din Gent a fost actualizată în raport cu versiunea anterioară publicată în 1996; noile criterii fac posibilă diagnosticarea sindromului Marfan pe bază clinică, fără teste genetice, cu sensibilitate și specificitate ridicate. Diagnosticul apare:
În absența unui istoric familial al sindromului Marfan:
- cu o rădăcină aortică Z-scor mai mare sau egal cu 2,00 ȘI ectopia lentis sau
- cu un scor Z al rădăcinii aortice mai mare sau egal cu 2,00 ȘI o mutație a genei FBN1 sau
- cu un scor Z de rădăcină aortică mai mare sau egal cu 2,00 ȘI un scor sistemic de 7 sau mai multe puncte sau
- cu ectopia lentis ȘI o mutație a genei FBN1 în prezența unei patologii aortice documentate.
Dacă aveți antecedente familiale de sindrom Marfan:
- Ectopia lentis sau
- Scor sistemic de 7 sau mai multe puncte sau
- Scorul Z al rădăcinii aortice mai mare sau egal cu 2,00.
Scorul sistemic în cauză poate fi calculat după cum urmează:
- Marca degetului mare ȘI marca pulsului: 3 puncte (marca degetului mare SAU marca mâinii: 1 punct).
- Piept carinat: 2 puncte (pectus excavatum sau altă asimetrie osoasă toracică: 1 punct).
- Deformitatea piciorului posterior la nivelul talusului și / sau calcaneului : 2 puncte ( picior plat : 1 punct).
- Ectazia durală : 2 puncte.
- Protrusio acetabuli : 1 punct.
- Pneumotorax spontan: 2 puncte.
- Raportul dintre segmentul superior și segmentul inferior al corpului mai mic de 0,87 ȘI raportul dintre lățimea brațului și înălțimea mai mare de 1,05 ȘI absența scoliozei severe: 1 punct.
- Scolioză toraco-lombară și / sau cifoză : 1 punct.
- Extinderea cotului în răpire mai mică de 170 de grade: 1 punct.
- Prezența a cel puțin 3 din următoarele 5 anomalii craniofaciale: enoftalmie , dolichocefalie , pleoapele în jos, hipoplazie malară , retrognatie : 1 punct.
- Vergeturile : 1 punct.
- Miopie mai mare de 3 dioptrii : 1 punct.
- Prolapsul valvei mitrale : 1 punct.
Diagnostic diferentiat
Sindromul Marfan intră în diagnostic diferențial cu alte boli ale țesutului conjunctiv și alte boli genetice:
- Sindromul Beals , care prezintă arahnodactilie , cifoscolioză și ectazie a aortei ascendente, dar prezintă contracturi articulare congenitale nedureroase, cu camptodactilie și hipotonie generalizată medie-severă, care sunt mai puțin frecvente în sindromul Marfan.
- Fenotip MASS , care împărtășește anomaliile scheletice, articulare și ale valvei cardiace ale sindromului Marfan, dar nu are o boală aortică care progresează cu aceeași severitate.
- Sindromul Stickler , care poate da anomalii cranio-faciale, detașarea retinei , miopie severă și alte simptome prezente în sindromul Marfan, dar, în general, dă o implicare auriculară importantă cu surditate progresivă, ceea ce nu se întâmplă în sindromul Marfan.
- Sindromul Shprintzen-Goldberg , o boală foarte rară diagnosticată doar în ultimele decenii; implică anomalii faciale mai grave decât cele ale sindromului Marfan și, mai presus de toate, este foarte des însoțită de întârziere mintală .
- Sindromul Ehlers-Danlos , care provoacă hipermobilitate articulară chiar mai mare decât cea a sindromului Marfan, modificări ale pielii și alte simptome tipice defectelor congenitale ale țesutului conjunctiv; forma vasculară a sindromului Ehlers-Danlos poate da, de asemenea, anevrisme de disecție nu numai aortei, ca în cazul sindromului Marfan, ci și multor alte artere care în cazul sindromului Marfan sunt rareori modificate.
- Homocisteinuria , care cauzează defecte scheletice similare cu cele ale sindromului Marfan (în special pieptul cu cheile, cifoscolioza, varusul piciorului ) și defecte oculare particulare, cum ar fi ectopia lentis și cataracta , dar foarte adesea implică o întârziere mintală care, la subiecții Marfan, are aceeași prevalență ca și în populația generală.
- Neoplazie endocrină multiplă de tip 2B , care conferă un aspect "marfanoid" persoanelor afectate (statură înaltă, subțire excesivă, membre conice, hipoplazie musculară ușoară).
- Sindromul Loeys-Dietz , care implică și ectazia durală, dar se distinge de sindromul Marfan prin uvula bifidă frecventă, alte anomalii ale palatului , anomalii în vindecarea rănilor și hipertelorism ocular.
- Sindromul Klinefelter , care, la fel ca sindromul lui Marfan, determină membrele foarte lungi în comparație cu trunchiul și statura înaltă; deși nu are multe semne comune cu sindromul Marfan, fiind o boală relativ frecventă, este adesea diagnosticată greșit în locul sindromului Marfan însuși.
Terapie
Singura strategie utilizată pentru a încetini slăbirea și extinderea aortei este utilizarea medicamentelor pentru a reduce tensiunea arterială. Beta-blocantele sunt de obicei prescrise, dar în ultima perioadă practica clinică pare să prezinte un beneficiu mai mare cu sartani ( losartan , telmisartan etc.) și cu inhibitori ai ECA . Sunt în curs studii pentru a se stabili eficacitatea sa efectivă. În special, a apărut, în timpul unui studiu pe un număr mic de pacienți cu sindrom Marfan, că perindoprilul ar fi deosebit de eficient. Studiul realizat la Baker Heart Institute din Melbourne a arătat o scădere semnificativă a diametrului aortic, rigiditatea aortei și o creștere consecventă a elasticității cu un risc mai mic de rupere. [26]
Au fost publicate rezultatele unui studiu „mic”, dar bine organizat, care arată pentru perindopril, verapamil și atenolol o reducere semnificativă a presiunii sistolice periferice și centrale, dar numai beta-blocante , care încetinesc ritmul cardiac și întârzie valul. Aortic, poate avea un rol constant în reducerea întârzierii expansiunii în arcada și aorta abdominală [27] . Cercetările au arătat că sartanii , în special losartanul , tind să încetinească semnificativ dilatarea aortei ascendente [28] , dar studiile ulterioare nu au confirmat această observație. [29]
Terapia chirurgicală (care implică o sternotomie și ulterior o înlocuire a porțiunii aortice afectate de anevrism, în timpul căreia folosim utilaje pentru circulația extracorporală ) vizează identificarea „calendarului chirurgical” potrivit pentru corectarea dilatației aortice înainte de acel nivel periculos. sunt atinse, ceea ce poate duce la disecție acută aortică . Studiile au arătat că un diametru aortic de 50 mm poate fi considerat tăiat pentru a interveni într-un timp relativ sigur. [30] . Alte studii mai recente, având în vedere un risc intraoperator scăzut pentru înlocuirea rădăcinii aortice, consideră în schimb 45 mm drept limita de diametru aortic dincolo de care se recomandă operația. Foarte des, în timpul acestor operații, o proteză non-organică Dacron este utilizată pentru a înlocui partea aortei afectată de dilatație anevrismală, uneori conform operației lui Bentall - efectuată pentru prima dată în 1968 și mai larg de la sfârșitul anilor șaptezeci [ 31] - care implică înlocuirea supapei aortice cu o supapă mecanică, uneori conform intervenției lui David care implică păstrarea supapei aortice naturale - deseori nu este practicabilă în cazul înrăutățirii insuficienței supapei. După prima intervenție chirurgicală este necesară urmarea terapiei anticoagulante orale pe viață și există în orice caz un risc tromboembolic crescut, în timp ce a doua tehnică chirurgicală nu necesită terapie anticoagulantă ulterioară, dar expune la un risc mai mare de a fi supus unei noi intervenții (acest risc este estimat la 1,3% pe an).
Mortalitatea peri-operatorie este mult mai mare (chiar și peste 10% în decurs de 30 de zile de la operație) în cazul intervenției chirurgicale de urgență, cu disecție acută în curs, decât în cazul chirurgiei elective (fără contextul de urgență al disecției), pentru care Mortalitatea pe 30 de zile scade la aproximativ 1,5%. [32]
Prognoză
Până la începutul anilor 1970, speranța medie de viață pentru pacienții cu Marfan era de aproximativ 40 de ani pentru bărbați și cu câțiva ani mai mare pentru femei; moartea ar putea să apară și la o vârstă foarte fragedă, în esență din cauza disecției aortice și a consecințelor ruperii vasului, precum și din insuficiența cardiacă legată de agravarea condițiilor de prolaps mitral și insuficiență aortică . S-au făcut îmbunătățiri semnificative în acest sens; datorită unui stil de viață adecvat (evitarea sporturilor periculoase pentru pereții aortici și care cresc mult tensiunea arterială), posibilitatea intervenției chirurgicale pe aorta, inima și alte părți ale corpului, metode mai eficiente de diagnostic și screening și medicamente care ajută pentru a limita și a amâna principalele complicații ale bolii, speranța de viață - conform mai multor studii - nu diferă prea mult de cea a populației generale.
Altre ricerche vedono ancora un'ampia differenza nella mortalità dei pazienti Marfan rispetto ai soggetti sani: uno studio norvegese conclusosi nel 2015 su una coorte di pazienti con sindrome di Marfan ha rilevato un'aspettativa di vita mediana pari a 63 anni per i maschi ea 73 anni per le femmine, con una mortalità per tutte le cause rispetto alla popolazione generale dello stesso sesso più alta di 8 volte negli uomini e di 4 volte nelle donne Marfan: pur essendo un dato in aumento rispetto a uno studio danese condotto negli anni '90, che vedeva un'età mediana di morte di 58 anni per i maschi Marfan e di 63 anni per le donne affette, esso mostra una differenza significativa rispetto alla sopravvivenza media della popolazione generale norvegese. I problemi cardiovascolari, in particolare a livello dell'aorta (dissezioni anche decenni dopo gli interventi chirurgici di sostituzione di parti dell'arteria), sembrano essere tuttora la causa più frequente di decesso tra i pazienti Marfan. [33]
Oltre a determinare una mortalità perioperatoria molto superiore, un intervento emergenziale di riparazione dell'aorta nel corso di una dissezione acuta sembra essere correlato ad tasso di sopravvivenza più basso anche sul lungo periodo: uno studio ha calcolato una sopravvivenza a 20 anni superiore al 70% nel caso di sostituzione della radice aortica a scopo preventivo, ma inferiore al 50% se lo stesso intervento è effettuato in condizioni di urgenza o di emergenza (dissecazione incipiente o acuta). [34]
Celebrità affette dalla sindrome
- John Tavener (1944-2013), compositore britannico . [35]
- Isaiah Austin (1993-), cestista statunitense . [36]
- Jonathan Jeanne (1997-), cestista francese . [37]
- Dominique de Roux (1935-1977), scrittore francese.
- Javier Botet (1977-), attore spagnolo .
- Troye Sivan (1995-), cantautore australiano .
- Peter Mayhew (1944-2019), attore britannico . [38]
- Bradford Cox (1982-), cantante statunitense. [39]
Nell'arte
MO (2007) è un film del regista statunitense Brian Scott Lederman , con protagonista Erik Per Sullivan , in cui si parla di un teenager che soffre della sindrome di Marfan. Il film è ispirato alla vita di Matthew Benjamin Lederman, fratello del regista, deceduto in giovane età a causa della sindrome.
L'attore spagnolo Javier Botet è noto per aver interpretato personaggi in film horror caratterizzati da deformità fisiche ottenibili senza effetti speciali solo grazie alle peculiarità specifiche della sindrome di Marfan. Tra questi, Slender Man e The Conjuring 2 .
Note
- ^ 17.4.4 Malattie del tessuto connettivo | SPREAD Live 2011
- ^ HUGO Gene Nomenclature Committee (HGNC), Symbol Report: FBN1 , su genenames.org . URL consultato il 26 lug 2017 (archiviato dall' url originale il 14 marzo 2018) .
- ^ Luigi Chiariello, "Sindrome di marfan, percorso diagnostico terapeutico assistenziale" ( PDF ), Fondazione Policlinico Tor Vergata, dicembre 2012. URL consultato il 26 lug 2017 (archiviato dall' url originale il 14 marzo 2018) .
- ^ ( EN ) Jane Kelly - updated : 14/07/2015, Marfan Syndrome; MFS , su omim.org , 6 giugno 2017. URL consultato il 26 luglio 2017 .
- ^ What causes Marfan syndrome? , su childrensnational.org .
- ^ a b c d e Sindrome di Marfan , su atlantemedicina.wordpress.com .
- ^ Disease-specific Growth Charts of Marfan Syndrome Patients in Korea
- ^ Pulmonary function in the Marfan syndrome
- ^ High prevalence of obstructive sleep apnea in Marfan's syndrome
- ^ Relation of aortic root dilatation and age in Marfan's syndrome
- ^ Aortic Event Rate in the Marfan Population
- ^ Zipes, Libby Bonow Braunwald (2005). Braunwald's Heart Disease ~ A Textbook of Cardiovascular Medicine, Seventh Edition. United States of America: Elseview Saunders. pag. 1894
- ^ Results of modern mitral valve repair in patients with Marfan syndrome
- ^ Observational Cohort Study of Ventricular Arrhythmia in Adults with Marfan Syndrome Caused by FBN1 Mutations
- ^ Giuseppe Canepa, Sindromi dismorfiche e malattie costituzionali dello scheletro , Padova, Piccin, 1996, p. 1947.
- ^ About Marfan Syndrome (in inglese)
- ^ Marfan Syndrome su omim.org (inglese)
- ^ Pulmonary involvement in patients with Marfan Syndrome
- ^ A case-control study of cutaneous signs in adult patients with Marfan disease: diagnostic value of striae
- ^ Coagulation Disorders in Marfan Syndrome (PDF)
- ^ Hemostatic abnormalities in adult patients with Marfan syndrome
- ^ Collagenopathies—Implications for Abdominal Wall Reconstruction: A Systematic Review
- ^ Neonatal Marfan syndrome
- ^ A Recurring FBN1 Gene Mutation in Neonatal Marfan Syndrome
- ^ A Case of Neonatal Marfan Syndrome: A Management Conundrum and the Role of a Multidisciplinary Team
- ^ Effect of perindopril on large artery stiffness and aortic root diameter in patients with Marfan syndrome: a randomized controlled trial
- ^ Effects of atenolol, perindopril and verapamil on haemodynamic and vascular function in Marfan syndrome – a randomised, double-blind, crossover trial
- ^ Losartan reduces aortic dilatation rate in adults with Marfan syndrome: a randomized controlled trial
- ^ Losartan Versus Atenolol for Prevention of Aortic Dilation in Patients With Marfan Syndrome
- ^ Cardiovascular SurgeryAortic Event Rate in the Marfan PopulationA Cohort Stud
- ^ The Button Bentall procedure
- ^ Replacement of the Aortic Root in Patients with Marfan's Syndrome
- ^ Survival, causes of death, and cardiovascular events in patients with Marfan syndrome
- ^ Replacement of the Aortic Root in Patients with Marfan's Syndrome
- ^ Morto John Tavener
- ^ Isaiah Austin has Marfan Syndrome (in lingua inglese)
- ^ Jeanne souffre de la mème maladie qu'Isaiah Austin (in francese)
- ^ Cenno alla sindrome di Marfan nella biografia di Mayhew su infarawaygalaxy.com
- ^ Cenno alla sindrome di Marfan nella biografia di Cox su AllMusic.com
Altri progetti
-
 Wikimedia Commons contiene immagini o altri file sulla sindrome di Marfan
Wikimedia Commons contiene immagini o altri file sulla sindrome di Marfan
Collegamenti esterni
- OMIM , su ncbi.nlm.nih.gov .
- Marfan Life Forum , su marfanlife.net . URL consultato il 14 gennaio 2009 (archiviato dall' url originale il 12 febbraio 2009) .
- Marfan Syndrome Forum [ collegamento interrotto ] , su forum.marfan-forum.org.uk .
- Marfan Friends World , su marfan-friends-world.org.uk . URL consultato il 30 giugno 2010 (archiviato dall' url originale il 30 marzo 2010) .
- Marfan Clinic , su marfanclinic.it .
- Marfan Bologna e Regione Emilia Romagna , su aosp.bo.it .
| Controllo di autorità | GND ( DE ) 4168872-7 |
|---|