Sindromul Von Hippel-Lindau
| Sindromul Von Hippel-Lindau | |
|---|---|
| Boala rara | |
| Cod. SSN | RN0780 |
| Specialitate | genetică clinică și neurologie |
| Clasificare și resurse externe (EN) | |
| ICD-9 -CM | 759,6 |
| OMIM | 193300 |
| Plasă | D006623 |
| eMedicină | 1219430 |
| GeneReviews | Prezentare generală |
| Eponime | |
| Arvid Lindau Eugen von Hippel | |
Sindromul VHL sau von Hippel-Lindau este o boală ereditară foarte rară caracterizată prin combinația diferitelor forme de neoplazie, inclusiv angioamele și alte forme de neoplazie renală și feocromocitoame .
Epidemiologie
Afectează una din 36.000 de persoane. Incidența este mai mare în jur de 30-40 de ani, deși poate apărea la orice vârstă.
fundal
Numele sindromului se datorează celor care l-au descris prima dată: oftalmologul german Eugen von Hippel și patologul suedez Arvid Lindau .
Etiologie
Cauza acestei boli este mutația unei gene situată pe brațul scurt al celui de-al treilea cromozom, cartografiat în 3p25-26, care codifică proteina VHL. Acesta din urmă colaborează în mod normal cu elonginele B și C [1] pentru a se lega de factorii de transcripție HIF (factorul indus de hipoxie) hidroxilați prin prezența oxigenului , provocând ubiquitinizarea și distrugerea lor prin proteazom . Complexul cu gene HIF este responsabil pentru răspunsurile celulare la hipoxie, inclusiv dezvoltarea neoangiogenezei și inducerea proliferării celulare; prin urmare, VHL se comportă ca un supresor tumoral și consecințele clinice ale lipsei sale sunt ușor deductibile. Produsul genetic poate fi complet absent ( deleții sau mutații framehift ) cu expresie anormală a codonilor STOP) sau inactiv, ducând la diferite imagini clinice.
De asemenea, blochează VEGF .
Simptomatologie
În funcție de genotipul subiectului, pot fi găsite diferite imagini clinice; în special, boala poate fi împărțită în 2 tipuri. Tipul 1 cu absența feocromocitomului și tipul 2 care în schimb îl manifestă. În primul caz, proteina este adesea mutată, dar prezentă; în al doilea caz, însă, este șters. În plus, tipul 2 este împărțit în continuare în 3 subtipuri: A, B și C (prima cu tumoră de celule renale, a doua cu hemangioblastom și a treia cu numai feocromocitom). Se mai găsesc tumori endocrine (în special pancreatice), chisturi renale (datorate edemului datorat neoangiogenezei) și alte manifestări neoplazice.
Diagnostic
Căutarea alterărilor genei VHL trebuie să fie ghidată de clinica cu care se prezintă boala și de buna practică clinică. (Trebuie evaluată utilitatea reală și cea mai potrivită metodă de consiliere genetică ). În cazul în care se suspectează o ștergere, se efectuează teste pentru a o găsi; în cazul unei mutații punctuale, se utilizează metode adecvate și se evaluează dacă mutația găsită corespunde unei patologii; sau consultând baze de date specifice sau efectuând teste indirecte (în tumorile subiectului se caută și ștergerea alelei sănătoase pe lângă cea mutată congenital, pentru a dovedi că dauna constatată este cea responsabilă pentru tabloul clinic) .
Terapie
Trecem la operație, care se efectuează în condiții particulare:
- Leziuni simptomatice ale coloanei vertebrale
- Leziuni cerebrale simptomatice sau cu evoluție rapidă
- Neoplasme renale solide> 3 cm
- Neoplasme pancreatice solide> 3 cm
Galerie de imagini
- Hemangioblastoamele în boala von Hippel-Lindau.




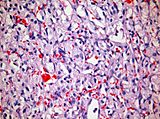
- Histopatologia hemangioblastoamelor.







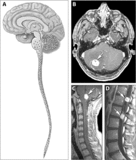
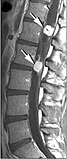
- Imagistica prin rezonanță magnetică a hemangioblastoamelor în boala von Hippel-Lindau.








Notă
- ^ Copie arhivată , la qs1439.pair.com . Adus pe 21 iunie 2013 (arhivat din original la 4 martie 2016) .
Bibliografie
- Tratat de chirurgie oncologică. Francesco Mazzeo, Pietro Forestieri. PICCIN, 2003. ISBN 978-88-299-1654-2
- „Principiile medicinii interne”, Harrison, McGraw-Hill, ediția a 15-a
Elemente conexe
Alte proiecte
-
 Wikimedia Commons conține imagini sau alte fișiere despre sindromul von Hippel-Lindau
Wikimedia Commons conține imagini sau alte fișiere despre sindromul von Hippel-Lindau
linkuri externe
- ( EN ) Sindromul Von Hippel-Lindau , în Encyclopedia Britannica , Encyclopædia Britannica, Inc.