Distrofia corneei
| Distrofia corneei | |
|---|---|
 | |
| Boala rara | |
| Cod. SSN | RFG140 |
| Specialitate | neurologie |
| Clasificare și resurse externe (EN) | |
| ICD-10 | H18.5 |
| Plasă | D003317 |
Distrofia corneei se referă la un grup eterogen de factori de mediu neinflamatori , progresivi, fără legătură care afectează corneea . Acestea sunt, de obicei, tulburări bilaterale și simetrice determinate genetic, care nu sunt asociate cu tulburări sistemice care provoacă opacificare cu reducerea consecventă a acuității vizuale [1] [2] [3] . Termenul de distrofie corneeană este imprecis, dar continuă să fie folosit pentru valoarea sa clinică [2], deși pentru mulți poate avea un sens mai istoric decât practic [1] [4] .
Distrofiile corneene au fost identificate la om, dar și la câini și, mai rar, la pisici.
Definiție și caracteristici
Termenul de distrofie corneeană a fost introdus în 1890 de Groenouw care a raportat cazul a 2 pacienți cu ceea ce el a numit noduli corneeni referindu-se probabil la o distrofie granulară și o distrofie maculară. Ulterior, Biber a numit distrofia corneei un caz de distrofie asemănătoare latexului. Mai târziu, termenul a fost folosit de Fuchs, Uhthoff și Yoshida. [1] Definiția este controversată și de unii termenul „distrofie” este considerat înșelător în unele cazuri, deoarece unele forme se manifestă mai degrabă ca degenerative decât bolile ereditare. Mai mult, chiar și la unii dintre cei a căror asociere cu un locus specific a fost identificat, gena nu a fost identificată sau caracterul moștenirii nu a fost înțeles (de obicei autozomal dominant sau recesiv ) [1] . Unele sunt asimptomatice sau unilaterale sau sunt asociate cu tulburări sistemice. Acestea sunt caracterizate de obicei printr-o alterare morfofuncțională rezultată din modificări ale trofismului corneean normal [3] .
Nomenclatură și clasificare
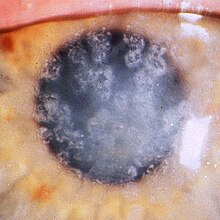
Natura eterogenă a tulburărilor, precum și utilizarea înșelătoare a termenului "distrofie corneeană" atribuită tulburărilor degenerative și tendința de a identifica și denumi noi forme de distrofie corneeană înainte de existența efectivă a unei noi boli a fost constatată și împărtășită, a generat multe neînțelegeri și, în unele cazuri, diagnostic incorect [1] [5] .
Pentru a soluționa controversele cu privire la clasificarea și nomenclatura distrofiilor corneene, s-a format Comitetul Internațional pentru Clasificarea Distrofiilor Corneale (IC3D) care în 2008 a propus o clasificare, care a fost apoi actualizată în 2015. Distrofiile corneene pot fi clasificate clinic în grupuri, pe baza asupra localizării anatomice a anomaliei. Unele afectează în principal epiteliul corneean, membrana bazală a acestuia sau stratul Bowman și stroma corneană superficială (distrofii corneene anterioare), stroma corneeană (distrofii stromale corneene) sau membrana lui Descemet și endoteliul cornean (distrofiile corneene) din spate) [4] . În analiza IC3D a fost depășită o clasificare prea dependentă de straturile corneene specifice, grupând distopiile corneene în epiteliale și subepiteliale, stromale, endoteliale. Diferitele distrofii care pot afecta diferite straturi, dar care depind de o singură genă (TGFBI) și locus sunt grupate într-un al patrulea grup numit epitelial-stromal [1] . Progresele recente în genetică moleculară au identificat multe dintre defectele genetice responsabile de majoritatea distrofiilor corneene [4] [6] , dar evoluția rapidă și continuă a informațiilor pe baza genetică a distrofiilor corneene, precum și a unor distrofii dependente de zeci de mutații [ 7] , a condus în 2015 la decizia I3CD de a nu specifica gena și locusul diferitelor distrofii.
| Clasificarea IC3D (2015) a distrofiilor corneene [1] | Baza genetică [1] [4] | ||||
|---|---|---|---|---|---|
| grup | Distrofia corneei | Categorie | Moştenire | Locus | Gene |
| Epitelial și subepitelial | |||||
| membrana bazală epitelială (EBMD) | 1 în cazuri rare | mai ales degenerative, sporadice | 5q31 | TGFBI în cazuri rare | |
| de eroziuni epiteliale recurente (ERED) și variante: Franceschetti (FRCD), Smolandiensis (DS), Helsinglandica (DH). | 4-3 | Autosomal dominant | Străin | Străin | |
| Mucinoasa subepitelială (SMCD) | 4 | Autosomal dominant | Străin | Străin | |
| de Meesmann (MECD) | 1 | Autosomal dominant | 12q12, 17q12 | KRT3, KRT12 | |
| Epiteliul Lisch (LECD) | 2 | cromozomul X dominant | Xp 22.3 | Străin | |
| Picătură gelatinoasă (GDCD) | 1 | Autosomal recesiv | Ip32 | TACSTD2 | |
| TGFBI epitelial-stromal | |||||
| de Reis-Bucklers (RBCD) | 1 | Autosomal dominant | 5q31 | TGFBI | |
| de Thiel-Benke (TBCD) | 2 | Autosomal dominant | 10q24 | Străin | |
| Reticular (latex), tip I (LCDI) | 1 | Autosomal dominant | 5q31 | TGFBI | |
| varianta (III, IIIA, I / IIIA, IV) a LCDI | 1 | Autosomal dominant | 5q31 | TGFBI | |
| Granular tip I (GCDI) | 1 | Autosomal dominant | 5q31 | TGFBI | |
| Granular tip II (GCD2) - de la Avellino | 1 | Autosomal dominant | 5q31. | TGFBI | |
| Stromal | |||||
| Macular (MCD) | 1 | Autosomal recesiv | 16Q22 | CHST6 | |
| de Schnyder (SCCD) | 1 | Autosomal dominant | 1p36 | UBIAD1 | |
| Stromal congenital (CSCD) | 1 | Autosomal dominant | 12q31.33 | DCN | |
| de Fleck (FCD) | 1 | Autosomal dominant | 2q35 | PIP5K3 | |
| Amorf posterior (PACD) | 3 | Autosomal dominant | Străin | Străin | |
| a Nebuloasei centrale Francois (CCDF) | 4 | Necunoscut | Străin | Străin | |
| Pre-Descemetic | 1-4 | Necunoscut | Străin | Străin | |
| Endotelial | |||||
| de Fuchs (FECD) cu debut tardiv | 1-2-3 | Necunoscut, uneori autosomal dominant | 13pter-q12.13 (FECD2), 18q21.2-q21.3 (FECD3), 20p13-p12 (FECD4), 5q33.1-q35.2 (FECD5), 10p11.2 (FECD6), 9p24.1-p22.1 (FECD7), 15q25 (FECD8). | Necunoscut, TCF8, SLC4A11 | |
| Boala Fuchs (FECD) cu aspect precoce | 1 | Autosomal dominant | Ip34.3 | COL8A2 | |
| Polimorf posterior (PPCD) 1 | 2 | Autosomal dominant | 20p11.12-q11.2 | Străin | |
| Polimorf posterior (PPCD) 2 | 1 | Autosomal dominant | Ip34.3-p32.3 | COL8A2 | |
| Polimorf posterior (PPCD) 3 | 1 | Autosomal dominant | 10p11.12 | ZEB1 | |
| Endotelial congenital ereditar (CHED) | 2 | Autosomal dominant | 20p11.12-q11.2 | Străin | |
| Endotelial legat de X (XECD) | 2 | cromozomul X dominant | Xq25 | Străin | |
| Legenda : - Categoria 1 : distrofie bine definită, din care o genă responsabilă specifică a fost cartografiată și identificată - Categoria 2 : distrofie bine definită, din care o genă a fost cartografiată în unul sau mai mulți loci cromozomiali specifici, dar alte gene sunt încă nu a fost identificat - Categoria 3 : distrofie bine definită, ale cărei gene responsabile nu au fost încă cartografiate - Categoria 4 : distrofie nouă sau documentată în trecut, în care dovezile că este o entitate distinctă nu sunt pe deplin convingătoare [1] - TGFBI : gena beta factor factor de creștere indus. | |||||
Distrofiile corneene de categoria 4, pe măsură ce apar noi dovezi care le pot distinge de alte distrofii, pot trece la o categorie inferioară. În caz contrar, acestea pot fi anulate, după cum a decis IC3D pentru distrofia Grayson-Wilbrandt [1] [8] .
Diagnostic
Suspiciunea de distrofie a corneei apare în cazul pierderii transparenței corneei sau a opacificării spontane a corneei, mai ales dacă este bilaterală și dacă există alți subiecți afectați în familie. Diagnosticul clinic se bazează pe vârsta de debut și aspectul clinic al corneei sub biomicroscopul cu lampă cu fantă. Pentru a defini tipul specific de distrofie sau degenerare, este necesară examinarea cu microscopie ușoară și microscopie electronică de transmisie (TEM) a țesutului corneean. În subtipurile de distrofii despre care se știe că au o mutație genetică, se poate efectua o analiză moleculară pentru a confirma diagnosticul [2] .
Diagnostic diferentiat
În absența testelor genetice și a elementelor de fond (familiaritate) diagnosticul diferențial poate fi complex, trebuind să se compare cu gammopatiile monoclonale, amiloidoză , deficit de lecitină-colesterol-aciltransferază, boala Fabry , cistinoză , tirozinemie tip 2, boli sistemice datorate stocării lizozomale (mucoplizaharidoză, lipidoză, mucolipidoză) și diverse boli ale pielii ( ihtioză legată de X , transferând cheratoza spinulară foliculară) [2] .
Notă
- ^ a b c d e f g h i j Weiss Jayne S.; Møller Hans Ulrik, Aldave, Anthony J., Seitz Berthold, Bredrup Cecilie, Kivelä Tero, Munier Francis L., Rapuano Christopher J., Nischal Kanwal K., Kim Eung Kweon, Sutphin John, Busin Massimo, Labbé Antoine, Kenyon Kenneth R ., Kinoshita Shigeru, Lisch Walter, Clasificarea IC3D a distrofiilor corneene - Ediția 2 , vol. 34, nr. 2, februarie 2015.
- ^ a b c d ORFANET: Distrofia corneei
- ^ a b Unitatea Clinică Operativă Oftalmologie CLINICĂ - Friuli Veneția Giulia - DISTROFE CORNEALE EROITARE
- ^ a b c d Gordon K Klintworth, Distrofiile corneene , în Orphanet Journal of Rare Diseases , vol. 4, nr. 7, februarie 2009.
- ^ Weiss JS., Distrofie cristalină Schnyder sinusoidală. Recomandare pentru o revizuire a nomenclaturii. , în Oftalmologie , vol. 103, 1996, pp. 465–473.
- ^ N.Pescosolido, M.Autolitano - Distrofii corneene
- ^ IC3D (2008): Tabelul genelor și mutațiilor asociate cu distrofiile corneene
- ^ ORPHANET: Distrofia corneei Grayson-Wilbrandt
Elemente conexe
Alte proiecte
-
 Wikimedia Commons conține imagini sau alte fișiere despre distrofia corneei
Wikimedia Commons conține imagini sau alte fișiere despre distrofia corneei

| Controlul autorității | Tezaur BNCF 53659 |
|---|