Molecula diatomică

O moleculă diatomică este o moleculă formată din doi atomi ; prin urmare, ele constituie cele mai simple forme de compuși moleculari care există. Moleculele diatomice sunt împărțite în molecule homonucleare , compuse din atomi ai aceluiași element chimic , și heteronucleare , compuse din atomi din diferite elemente.
Molecule homonucleare
O moleculă diatomică homonucleară este o moleculă formată din doi atomi egali.
Unele elemente ale tabelului periodic au o stare standard sub forma unei molecule diatomice, ca în cazul hidrogenului (H 2 ), azotului (N 2 ), oxigenului (O 2 ), fluorului (F 2 ), clorului (Cl 2 ), brom (Br 2 ) și iod (I 2 ). Fosforul (P 4 ) și sulful (S 8 ) nu sunt diatomice
Molecula H 2 +


Moleculele diatomice homonucleare sunt compuse din doi atomi ai aceluiași element chimic; cea mai simplă dintre acestea este H 2 + , pentru care ecuația electronică ia forma: [1]
![\ left [{\ frac {\ hbar ^ 2} {2m_e}} \ nabla_r ^ 2 - \ frac {ke ^ 2} {| \ mathbf {r} + \ mathbf {R} / 2 |} - \ frac {ke ^ 2} {| \ mathbf {r} - \ mathbf {R} / 2 |} + \ frac {ke ^ 2} {R} \ right] \ psi _ {\ mathrm {e}} (\ mathbf {r} ) = E _ {\ mathrm {e}} (\ mathbf {r}) \ psi _ {\ mathrm {e}} (\ mathbf {r})](https://wikimedia.org/api/rest_v1/media/math/render/svg/dbdcc15a178e5f1487ae8efb1746b1a94d4898bc)
unde este , al doilea și al treilea termen reprezintă atracția V ne a electronului către nuclei și al patrulea repulsia celor doi nuclei.
Cei doi protoni formează două puțuri potențiale, iar funcția de undă electronică este combinația liniară a două funcții de undă asemănătoare hidrogenului : [2]


![\ psi _ {\ mathrm {\ pm}} (\ mathbf {r}) = \ frac {1} {\ sqrt {2}} [\ psi_ {1s} (\ mathbf {r} + \ mathbf {R} / 2) \ pm \ psi_ {1s} (\ mathbf {r} - \ mathbf {R} / 2)]](https://wikimedia.org/api/rest_v1/media/math/render/svg/fdf6c56809c63e4c6bfd05f1afb629af6a11f059)
Funcția de undă constituie legătura moleculară orbitală , funcția constituie orbitalul anti - legătură . [3] Orbitalul de legare are o energie mai mică decât orbitalul anti-legătură și, prin urmare, este cel mai probabil.
Funcții , deși descriu bine distribuția probabilității electronului în starea fundamentală, nu sunt soluții exacte ale ecuației electronice.



Funcția de undă , în spațiul dintre cele două nuclee, este mai mare decât funcțiile individuale de undă de tip hidrogen , și acest fapt generează legătura covalentă între cei doi nuclei. De fapt, se observă că densitatea de probabilitate asociată cu funcția de undă:

![| \ psi _ {\ mathrm {\ pm}} | ^ 2 = \ frac {1} {2} [\ psi_ {1s} ^ 2 (\ mathbf {r} + \ mathbf {R} / 2) + \ psi_ {1s} ^ 2 (\ mathbf {r} - \ mathbf {R} / 2) \ pm 2 \ psi_ {1s} (\ mathbf {r} + \ mathbf {R} / 2) \ psi_ {1s} (\ mathbf {r} - \ mathbf {R} / 2)]](https://wikimedia.org/api/rest_v1/media/math/render/svg/50a75eda255a84496516016b11905999d90085a1)
conține un termen de interacțiune, produsul dublu, care reprezintă suprapunerea celor două funcții de undă: este o regiune de sarcină negativă care unește cei doi nuclei de sarcină opusă.
În ceea ce privește orbitalul anti-legătură , dispare la mijloc între cele două nuclee, unde generează o densitate de probabilitate mai mică decât ar avea fără termenul de suprapunere.
Molecula H 2


Acum luați în considerare molecula H 2 , cea mai simplă moleculă neutră. Având doi electroni, funcția de undă electronică singlet este dată de: [4]
![\ psi _ {\ mathrm {S}} (1,2) = \ frac {1} {\ sqrt {2}} [\ psi_ {1s} (\ mathbf {r_1} - \ mathbf {R} / 2) \ psi_ {1s} (\ mathbf {r_2} + \ mathbf {R} / 2) + \ psi_ {1s} (\ mathbf {r_2} - \ mathbf {R} / 2) \ psi_ {1s} (\ mathbf {r_1 } + \ mathbf {R} / 2)] \ chi ^ A (1,2)](https://wikimedia.org/api/rest_v1/media/math/render/svg/909792d0f57537b94dea0a27378c910077e1d41c)
și reprezintă legătura orbitală, în timp ce cea a tripletului din: [5]
![\ psi _ {\ mathrm {T}} (1,2) = \ frac {1} {\ sqrt {2}} [\ psi_ {1s} (\ mathbf {r_1} - \ mathbf {R} / 2) \ psi_ {1s} (\ mathbf {r_2} + \ mathbf {R} / 2) - \ psi_ {1s} (\ mathbf {r_2} - \ mathbf {R} / 2) \ psi_ {1s} (\ mathbf {r_1 } + \ mathbf {R} / 2)] \ chi ^ S (1,2)](https://wikimedia.org/api/rest_v1/media/math/render/svg/efa707e0f683775b3cc7ef364bdf94eb4370d291)
reprezentând orbitalul anti-legătură, unde:

Și

sunt stările de rotire , unde + reprezintă rotirea, - rotirea.
Densitatea probabilității spațiale este: [5]
![{\ displaystyle | \ psi _ {\ mathrm {S, T}} | ^ {2} = {\ frac {1} {2}} [\ psi _ {1s} ^ {2} (\ mathbf {r_ {1 }} - \ mathbf {R} / 2) \ psi _ {1s} ^ {2} (\ mathbf {r_ {2}} + \ mathbf {R} / 2) + \ psi _ {1s} ^ {2} (\ mathbf {r_ {2}} - \ mathbf {R} / 2) \ psi _ {1s} ^ {2} (\ mathbf {r_ {1}} + \ mathbf {R} / 2) \ pm 2 \ psi _ {1s} (\ mathbf {r_ {1}} - \ mathbf {R} / 2) \ psi _ {1s} (\ mathbf {r_ {1}} + \ mathbf {R} / 2) \ psi _ {1s} (\ mathbf {r_ {2}} - \ mathbf {R} / 2) \ psi _ {1s} (\ mathbf {r_ {2}} + \ mathbf {R} / 2)]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/0d0321d97d2ee4d9fd4e369101f55dbd777cacec)
De asemenea, în acest caz, termenul de interferență reprezintă suprapunerea funcțiilor de undă de tip hidrogen în regiunea dintre nuclee și implică o creștere a sarcinii în cazul singletului (semnul +) și o scădere a sarcinii în triplet (- semn).
Molecule diatomice homonucleare din prima și a doua perioadă
Moleculele diatomice homonucleare din prima și a doua perioadă (H-Ne) au fost studiate pe larg.
Toate sunt molecule diamagnetice (S = 0), cu excepția B 2 și O 2 care sunt paramagnetice (S = 1). următorul tabel arată pentru fiecare moleculă termenul stării fundamentale, valoarea energiei de disociere (D e ) și distanța de echilibru (R e ).
Orbitalele moleculare care sunt umplute succesiv sunt obținute prin combinații liniare adecvate de orbitali atomici de tip 1s și 2s (σ g 1s / 2s și σ u * 1s / 2s), 2px și 2py (π u și π g * ) și 2pz (σ g 2p și σ u * 2p).
| Moleculă | Termen de stat fundamental | D e (eV) | R și (A) |
|---|---|---|---|
| H 2 | 1 Σ + g | 2,79 | 1,06 |
| El 2 | 1 Σ + g | 0,0009 | 3.00 |
| Li 2 | 1 Σ + g | 1,07 | 2,67 |
| Fii 2 | 1 Σ + g | 0,10 | 2.45 |
| B 2 | 3 Σ - g | 3.1 | 1,59 |
| C 2 | 1 Σ + g | 6.3 | 1.24 |
| N 2 | 1 Σ + g | 9,92 | 1.10 |
| SAU 2 | 3 Σ - g | 5.21 | 1.21 |
| F 2 | 1 Σ + g | 1,66 | 1,41 |
| Ne 2 | 1 Σ + g | 0,0036 | 3.1 |
Molecule diatomice homonucleare din a treia perioadă (Na-Cl)
Moleculele diatomice din a treia perioadă au fost caracterizate experimental la nivel spectroscopic. Cl 2 este starea standard pentru clorul elementar.
| Moleculă | Termen de stat fundamental | D e (eV) | R și (A) |
|---|---|---|---|
| Na 2 | 3.0788 | ||
| Mg 2 | 3.890 | ||
| La 2 | 2.466 | ||
| Da 2 | 2.246 | ||
| P 2 | 1.893 | ||
| S 2 | 1.889 | ||
| Cl 2 | 1,987 |
Molecule heteronucleare
La moleculele heteronucleare simetria care a caracterizat moleculele homonucleare lipsește, iar orbitalele nu sunt o combinație pură simetrică și antisimetrică a orbitalilor atomici. În astfel de molecule, orbitalele pot fi aproximate cu stările proprii ale unei matrice pătrate de dimensiunea 2: [6]

unde este:

este hamiltonianul eficient al electronului unic în timp ce stările Și sunt orbitalele corespunzătoare atomului stâng și respectiv al celui drept.
Valorile proprii asociate matricei sunt:



Orbitalii de legare și anti-legare sunt date de statele proprii:



cu:

pentru obținem molecula homonucleară și termenul reprezintă împărțirea între orbitalul de legare și antiblocarea unei molecule homonucleare sau împărțirea între combinații simetrice și antisimetrice. [6]
Dupa cum statele proprii de legare și anti-legătură seamănă din ce în ce mai mult cu orbitalii Și a atomilor individuali și același lucru se întâmplă pentru valorile proprii energetice respective. [7] Când diferența este de așa natură încât implică un transfer complet de sarcină între cei doi atomi, legătura se spune că este ionică .




Mișcări interne

Aproximarea Born-Oppenheimer , numită și aproximare adiabatică , ne permite să considerăm mișcarea nucleelor independentă de cea a electronilor , deoarece primii sunt extrem de încet și mai grei decât cei din urmă. Acest lucru face posibilă factorizarea funcției de undă totală a moleculei: [8] [9]

unde indicele e indică funcția de undă a electronilor, indicele n al nucleelor și Și sunt pozițiile nucleilor și respectiv ale electronilor.
Funcția de undă a electronilor, în aproximarea adiabatică, satisface ecuația valorii proprii:


![\ left [T_ \ mathrm {e} + V_ \ mathrm {ne} (\ mathbf {R}, \ mathbf {r}) + V_ \ mathrm {ee} (\ mathbf {r}) \ right] \ psi _ { \ mathrm {e}} (\ mathbf {R}, \ mathbf {r}) = E _ {\ mathrm {e}} (\ mathbf {R}) \ psi _ {\ mathrm {e}} (\ mathbf { R}, \ mathbf {r})](https://wikimedia.org/api/rest_v1/media/math/render/svg/ad4ab644a0a73006f448d26d3dea2c5b0e598731)
În timp ce potențialul care conduce mișcarea nucleelor:

se numește potențial adiabatic și stă la baza dinamicii moleculei.
Din expresia potențialului adiabatic este clar că dinamica nucleelor este condusă de energie furnizat de ecuația electronică: acest termen este fundamental, deoarece reprezintă „lipiciul” care ține împreună nucleii atomilor care alcătuiesc molecula. [10]
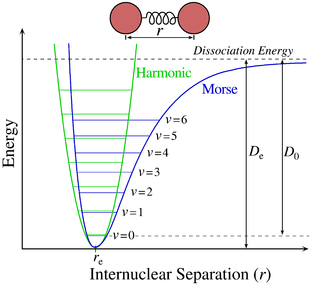
Pentru moleculele diatomice, potențialul adiabatic este un potențial armonic și poate fi aproximat de potențialul Morse , care, spre deosebire de oscilatorul armonic cuantic, include în mod explicit efectele ruperii legăturilor chimice , cum ar fi existența stărilor nelegate. Potențialul adiabatic este independent de poziția centrului de masă al moleculei și de orientarea liniei care unește cele două nuclee: se bucură, prin urmare, de invarianță în ceea ce privește translațiile și rotațiile, iar mișcarea nucleelor poate fi studiată ca problemă cu două corpuri . Pornind de la această abordare, ecuația Schrödinger poate fi separată în mișcare radială, în funcție de distanța dintre cei doi nuclei și mișcarea orbitală, în funcție de numărul cuantic orbital . Ecuația Schrödinger în cazul mișcării într-un câmp central este:

![\ left [- \ frac {\ hbar ^ 2} {2 (M + m)} \ nabla _ {\ mathbf r_ {cm}} ^ {2} - \ frac {\ hbar ^ 2} {2 \ mu} \ nabla ^ {2} + V_ \ mathrm {ad} (| \ mathbf r_ {rel} |) \ right] \ psi _ {\ mathrm {n}} (\ mathbf r_ {cm}, \ mathbf r_ {rel}) = E_ {tot} \ psi _ {\ mathrm {n}} (\ mathbf r_ {cm}, \ mathbf r_ {rel})](https://wikimedia.org/api/rest_v1/media/math/render/svg/2f63a966a7e40307058884854406f9068bf7029e)
unde este indică amplasarea centrului de masă e poziția relativă a celor două nuclee, diferența dintre pozițiile lor respective.
Problema poate fi apoi separată în două ecuații, una pentru centrul de masă și una pentru particula de masă μ care se deplasează într-un câmp central față de centrul de masă. Prin urmare, funcția de undă poate fi luată în considerare după cum urmează: . Ecuația pentru , care reprezintă problema particulelor libere , asigură energia de translație a moleculei. Ecuația pentru putem lua în calcul și partea radială, în funcție de r , și partea unghiulară, în funcție de coordonatele unghiulare: .
Soluția pentru sunt armonicele sferice , iar stările respective sunt stări proprii ale momentului unghiular orbital și ale componentei sale de-a lungul axei z .
Ecuația pentru în schimb, se spune : [11]









![\ left [- \ frac {\ hbar ^ 2} {2 \ mu} \ frac {d ^ 2} {dr ^ 2} + \ frac {\ hbar ^ 2 l (l + 1)} {2 \ mu r ^ 2} + V_ \ mathrm {ad} (| \ mathbf r_ {rel} |) \ right] g = E g](https://wikimedia.org/api/rest_v1/media/math/render/svg/a0d8b7c5343baef9266e16f45363c9237debc159)
unde al doilea termen reprezintă contribuția la energie de rotație , care depinde de numărul cuantic orbital l .
Potențialul adiabatic poate fi dezvoltat și în seria Taylor , care trunchiată la ordinea a doua este: [9]


unde este este valoarea care minimizează , și reprezintă poziția de echilibru a celor două nuclee. Această expresie reprezintă o mișcare armonică în jur care oferă o contribuție energetică dată de energia ecuației electronice conținută în și energia vibrațională .






Spus lungimea caracteristică dată de relație si a zis , soluțiile ecuației pentru Sunt:





unde este este polinomul hermit de grad .
Spectrul energetic conține în cele din urmă trei termeni:



Acești termeni sunt contribuțiile energetice care caracterizează dinamica moleculei diatomice și, în mod specific, sunt: [9] [12]
- Contribuția electronică, dată de termen din , care definește adâncimea a orificiului potențial generat de cele două nuclee, responsabile de legătura chimică. Nivelurile de energie asociate cu acest termen se numesc suprafețe adiabatice și corespund diferitelor stări de energie ale electronilor. Electronii care sunt promovați de la un orbital la altul, de exemplu de la un orbital de legătură la un orbital anti-legătură, tranziție între două valori Și a potențialului adiabatic. Aceste tranziții sunt de ordinul a 10 eV , iar diferite suprafețe adiabatice corespund, de asemenea, cu valori diferite ale . Tranzițiile electronice între două dintre aceste suprafețe sunt, de asemenea, însoțite de tranziții între diferite stări vibraționale și rotaționale.




- Contribuția vibrațională, mai puțin energică decât cea precedentă, decât în aproximarea mișcării armonice oferită de excluderea termenilor peste ordinea a doua în dezvoltarea anterioară a este dat de valorile proprii ale oscilatorului armonic cuantic :


- unde este este constanta lui Planck e frecvența unghiulară a oscilației din jur .
- Frecvența este dată de:



- cu

- Și masa redusă a oscilatorului cu două corpuri, dată de raportul dintre produs și suma maselor celor două nuclee.
- Această contribuție descrie mișcarea armonică a celor două nuclee în jurul poziției de echilibru, iar tranzițiile între două niveluri vibraționale sunt de ordinul unei zecimi din eV.

- Contribuția de rotație, cea mai puțin energică dintre cele trei, asigurată de ecuația unghiulară a atomului de hidrogen , egală cu:

- unde este este impulsul unghiular orbital e momentul inerției .
- Această contribuție este în general de ordinul meV și se calculează prin asumarea .



În concluzie, prin urmare, energia internă a unei molecule diatomice este:

unde termenii sunt enumerați în ordinea importanței.




Spectrul electromagnetic molecular
Spectrul electromagnetic molecular este generat de tranzițiile dintre două stări proprii ale energiei totale. În cazul studierii spectrului de emisie, molecula trece de la o stare excitată la starea fundamentală, în timp ce în cazul studierii spectrului de absorbție , se observă tranziția inversă. Acest pasaj implică emisia sau absorbția unui foton , a cărui frecvență este dată de legea lui Planck :

unde este este diferența de energie dintre cele două stări de plecare și sosire:


Tranzițiile electronice de la starea fundamentală la primele stări excitate sunt de ordinul câtorva eV și sunt observate în regiunea vizibilă și ultravioletă a spectrului electromagnetic , în timp ce tranzițiile roto-vibraționale sunt observate în regiunea infraroșie . [13]
Tranzițiile dintre două stări proprii ale energiei totale sunt studiate prin tranzițiile între stări proprii alemomentului dipol electric , definit ca: [9]

cu și încărcarea electronului.
Acest operator este explicat prin expresia:
![\ mathbf {d} = \ int {\ psi_ {vib} '^ * \ psi_ {rot}' ^ *} \ left [\ int \ psi_ {el} ^ * \ mathbf {d} \ psi_ {el} dx_e \ dreapta] \ psi_ {vib} \ psi_ {rot} d \ tau = \ langle {\ psi_ {vib} '\ psi_ {rot}'} | \ mathbf {\ mu} | \ psi_ {vib} \ psi_ {rot} \ rangle](https://wikimedia.org/api/rest_v1/media/math/render/svg/16e3a71f413c36ee9c61f3107e06dcdcd39d8a6a)
unde este este operatorul momentului dipolului de electroni al moleculei:



Fiecare dintre nivelurile vibraționale care caracterizează o suprafață adiabatică este asociat cu diferite stări de rotație. În diagrama spectroscopică, tranzițiile de rotație constituie două ramuri: prima se numește ramura R și reprezintă tranzițiile de rotație între numerele cuantice , în timp ce a doua, numită ramură P , reprezintă tranzițiile . Între cele două ramuri există un gol, motivat de faptul că tranziția este interzis de regulile de selecție. [14]



Când tranziția este realizată de un electron, generează, de asemenea, tranziții între stările proprii ale energiei roto-vibraționale a nucleelor: aceste tranziții sunt numite vibronice și sunt cauzate de faptul că geometriile diferite ale moleculei corespund două suprafețe adiabatice diferite. În special, în moleculele diatomice, acestea corespund diferitelor distanțe internucleare.
Spectrul nuclear
În cazul moleculelor diatomice homonucleare, momentul dipol electric este zero din motive de simetrie [15] și acest fapt explică transparența atmosferei Pământului , compusă în principal din O 2 și N 2 .
Pe de altă parte, în moleculele diatomice heteronucleare, elementul matrice al componentei de-a lungul axei z a momentului dipolar este: [9]


unde este sunt stări proprii simultane ale energiei vibraționale și rotaționale. La fel se întâmplă și pentru componentele x și y .
Din proprietățile armonicelor sferice și dezvoltarea în jurul distanței de echilibru se obțin regulile de selecție:



care definesc tranzițiile permise între stările proprii ale operatorului asociate cu dipolul electric observabil .

Spettro elettronico
Una transizione elettronica molecolare consiste in una transizione da parte dell'elettrone tra due superfici adiabatiche . Tali transizioni sono simili a quelle atomiche, e consistono nella promozione di un elettrone da un orbitale molecolare a un altro orbitale vuoto. [13]
Le regole di selezione si ricavano osservando che l'operatore di spin totale:

commuta con l'hamiltoniana elettronica e con , l'operatore di dipolo non agisce sullo spin, e pertanto si ha che . [9]
Per l'operatore di momento angolare nelle molecole biatomiche:



solo la componente lungo l'asse z commuta con , ottenendo che , mentre per le altre due componenti si ricava che . In definitiva si ha:




Il principio di Franck Condon
Il principio di Franck Condon afferma la probabilità associata a una transizione vibrazionale, data da:

aumenta all'aumentare della sovrapposizione delle funzioni d'onda dei rispettivi stati iniziale e finale. Questo comporta che i livelli vibrazionali associati allo stato finale sono favoriti nel momento in cui la transizione comporta un cambiamento minimo nelle coordinate nucleari. Una conseguenza del principio è che, ad esempio, come mostrato nella figura a sinistra, se le funzioni d'onda tra lo stato fondamentale della superficie adiabatica iniziale e il secondo stato eccitato della superficie adiabatica finale si sovrappongono, tale transizione è più probabile delle altre dal momento che minimizza la variazione delle coordinate dei nuclei.
Note
- ^ Brehm, Mullins , Pag. 503 .
- ^ Brehm, Mullins , Pag. 504 .
- ^ Brehm, Mullins , Pag. 507 .
- ^ Brehm, Mullins , Pag. 509 .
- ^ a b Brehm, Mullins , Pag. 510 .
- ^ a b Manini , Pag. 70 .
- ^ Manini , Pag. 71 .
- ^ Manini , Pag. 61 .
- ^ a b c d e f Renzo Cimiraglia - Note al corso di Spettroscopia Molecolare ( PDF ), su chim183.unife.it . URL consultato il 15 novembre 2010 (archiviato dall' url originale il 2 agosto 2007) .
- ^ Manini , Pag. 62 .
- ^ Brehm, Mullins , Pag. 523 .
- ^ Manini , Pag. 76 .
- ^ a b Manini , Pag. 79 .
- ^ Manini , Pag. 78 .
- ^ Brehm, Mullins , Pag. 528 .
Bibliografia
- ( EN ) John Brehm, William J. Mullins,Introduction To The Structure Of Matter: A Course In Modern Physics , Greenville, NC, USA, John Wiley & Sons, 1989, ISBN 978-0-471-60531-7 .
- ( EN ) Nicola Manini, Introduction to the Physics of Matter , Milano, CUSL, 2008, ISBN 978-88-8132-552-8 .
- Roberto Spinicci, Elementi di Chimica , Firenze, Firenze University Press, 2009, ISBN 978-88-6453-062-8 .
- ( EN ) Pauling, Linus, General Chemistry , New York, Dover Publications, Inc., 1970, ISBN 0-486-65622-5 .
- ( EN ) Ebbin, Darrell, D., General Chemistry, 3rd Ed. , Boston, Houghton Mifflin Co., 1990, ISBN 0-395-43302-9 .
- ( EN ) Brown, TL, Chemistry – the Central Science, 9th Ed. , New Jersey, Prentice Hall, 2003, ISBN 0-13-066997-0 .
- ( EN ) Chang, Raymond, Chemistry, 6th Ed. , New York, McGraw Hill, 1998, ISBN 0-07-115221-0 .
- ( EN ) Zumdahl, Steven S., Chemistry, 4th ed. , Boston, Houghton Mifflin, 1997, ISBN 0-669-41794-7 .
Voci correlate
Altri progetti
-
 Wikimedia Commons contiene immagini o altri file su molecola biatomica
Wikimedia Commons contiene immagini o altri file su molecola biatomica
Collegamenti esterni
- ( EN )Molecola biatomica , su Enciclopedia Britannica , Encyclopædia Britannica, Inc.