Fibroza pulmonară idiopatică
| Fibroza pulmonară idiopatică | |
|---|---|
| Specialitate | pneumologie |
| Clasificare și resurse externe (EN) | |
| OMIM | 178500 |
| Plasă | D054990 |
| MedlinePlus | 000069 |
| eMedicină | 363273 și 301226 |
Fibroza pulmonară idiopatică (IPF sau, fibroza pulmonară idiopatică) este o afecțiune cronică , invalidantă și fatală caracterizată printr-o scădere progresivă a funcției pulmonare. [1] [2] Termenul fibroză pulmonară înseamnă cicatrizarea țesutului și este cauza agravării dispneei (respirație scurtă). Fibroza este de obicei asociată cu un prognostic slab. [1] [2] [3] Termenul „ idiopatic ” este utilizat deoarece cauza fibrozei pulmonare este încă necunoscută. [1]
IPF apare de obicei la adulți cu vârste cuprinse între 50 și 70 de ani, în special la persoanele cu antecedente de fumat și afectează mai mult bărbații decât femeile. [1][4]
IPF aparține unui grup mare de peste 200 de boli pulmonare, cunoscute sub numele de boli pulmonare interstițiale (ILD), caracterizate prin afectarea interstițiului pulmonar. [2] Interstitiul, țesutul dintre alveolele din plămâni, este locul cel mai afectat de ILD. Cu toate acestea, aceste tulburări afectează adesea nu numai interstițiul, ci și spațiile aeriene, căile respiratorii periferice și vasele de sânge. [2] Țesutul pulmonar la persoanele cu IPF prezintă caracteristicile histopatologice ale pneumoniei interstițiale obișnuite (UIP). Prin urmare, UIP este omologul patologic al IPF. [1][4]
În 2011, au fost publicate noi orientări pentru diagnosticarea și gestionarea IPF. [1] Diagnosticul IPF necesită excluderea altor cauze cunoscute ale ILD și prezența caracteristicilor radiologice specifice, identificate prin tomografie computerizată de înaltă rezoluție (HRCT). Într-un cadru clinic adecvat, diagnosticul IPF poate fi pus prin utilizarea doar a HRCT, evitând necesitatea unei biopsii pulmonare chirurgicale. [1] [2]
Clasificare
Fibroza pulmonară idiopatică (IPF) aparține unui grup mare de peste 200 de boli pulmonare, cunoscute sub numele de boli pulmonare interstițiale (ILD), caracterizate prin afectarea interstitiului, [2] țesutul dintre alveolele din plămâni. IPF este o manifestare specifică a pneumoniei interstițiale idiopatice (IIP), care este ea însăși o formă de ILD, cunoscută și sub numele de boală interstițială pulmonară .
Clasificarea IIP-urilor publicată în 2002 de American Thoracic Society / European Respiratory Society (ATS / ERS) a fost actualizată în 2013. [2] Noua clasificare prevede subdiviziunea pneumoniei interstițiale idiopatice (IIP) în trei categorii principale: IIP primar , IIP rar și IIP neclasificabil. IIP-urile principale sunt grupate în:
- IP cu fibroză cronică (inclusiv IPF și pneumonia interstițială nespecifică [NSIP]),
- IP legat de fumat (de exemplu bronșiolită respiratorie asociată cu boli pulmonare interstițiale [RB-ILD] și pneumonie interstițială descuamativă [DIP])
- IP acută / subacută (de exemplu, pneumonie criptogenă organizatoare [COP] și pneumonie interstițială acută [AIP]). [2]
Diagnosticul IIP necesită excluderea cauzelor cunoscute ale ILD. Câteva exemple de ILD cauză cunoscută sunt pneumonita de hipersensibilitate , histiocitoza celulară Langerhans pulmonară , azbestoză și colagenopatia vasculară . Cu toate acestea, aceste boli afectează adesea nu numai interstițiul, ci și spațiile aeriene, căile respiratorii periferice și vasele de sânge. [2]
Figura de mai jos prezintă noua clasificare a IIP-urilor.

Epidemiologie
Deși rar, IPF este cea mai comună formă de IIP. [2] Prevalența IPF a fost estimată a fi între 14 și 42,7 la 100.000 de persoane, pe baza unei analize SUA a datelor colectate de asigurările de sănătate, cu unele diferențe pe baza definițiilor de caz utilizate în această analiză.[4] [5] IPF este mai frecvent la bărbați decât la femei și este de obicei diagnosticat la persoanele cu vârsta peste 50 de ani. [1] Este dificil să se determine incidența IPF, deoarece există o lipsă de aplicare uniformă a criteriilor de diagnostic. [1] [5] În cele 28 de țări ale Uniunii Europene, o serie de surse estimează o incidență asupra populației de 4,6-7,4 persoane la 100 000, [6][7] valoare care indică faptul că în fiecare an vor fi diagnosticați 30 000 –35.000 de cazuri noi de IPF.[4] [8]
Un studiu observațional recent, retrospectiv, de cohortă, care a inclus pacienți diagnosticați accidental cu ILD între 2003 și 2009 la Spitalul Universitar Aarhus din Danemarca a arătat că incidența ILD a fost de 4,1 la 100.000 de locuitori / an. IPF a fost cea mai frecvent diagnosticată boală (28%), urmată de ILD legată de boala țesutului conjunctiv (14%), pneumonită de hipersensibilitate (7%) și pneumonie interstițială nespecifică (NSIP) (7%). Incidența IPF a fost de 1,3 la 100.000 de locuitori / an. [9]
Datorită distribuției inegale a bolii în Europa, este necesar să se mențină o actualizare constantă a datelor epidemiologice printr-un registru european pentru ILD și IPF.
Cauze / Factori de risc ai IPF
Fibroza pulmonară idiopatică sau FPI este prin definiția sa idiopatică (ceea ce înseamnă că nu are o cauză cunoscută), cu toate acestea, s-a demonstrat că unii factori de mediu și expunerea la anumiți agenți cresc riscul de apariție a FPI. [10] Fumatul este cel mai recunoscut și acceptat factor de risc pentru IPF, crescând riscul de îmbolnăvire cu aproximativ dublu. [10] Unele expuneri de mediu, cum ar fi expunerea la metal, lemn, cărbune, silice, praf de piatră și expuneri profesionale legate de agricultură / creșterea animalelor, s-au dovedit, de asemenea, că cresc riscul de FPI. [11] Există unele dovezi că infecțiile virale sunt asociate cu fibroza pulmonară idiopatică și alte boli pulmonare fibrotice. [11]
Etiologie și patogenie
Deși IPF a fost studiat pe larg, cauzele sale sunt încă necunoscute. [1] Fibroza IPF a fost corelată cu fumatul țigării, factori de mediu (de exemplu, factori ocupaționali, cum ar fi expunerea la gaze, fum, substanțe chimice sau praf), alte afecțiuni medicale, inclusiv boala de reflux gastroesofagian (GERD) sau de la o predispoziție (formă familială). Cu toate acestea, nu toți acești factori sunt prezenți la toți indivizii cu IPF și, prin urmare, nu oferă o explicație satisfăcătoare pentru această patologie. [1] [12]
IPF este considerat a fi rezultatul unei alterări a procesului de vindecare a rănilor, care include și duce la depunere anormală și excesivă de colagen (fibroză) în interstitiul pulmonar cu inflamație minimă asociată. [13]
Se presupune că în IPF există leziuni inițiale sau repetate ale celulelor pulmonare, numite celule epiteliale alveolare (AEC), care ocupă o mare parte a suprafeței alveolare. [14] Când AEC de tip I sunt deteriorate sau distruse, se crede că AEC de tip II proliferează pentru a acoperi membrana bazală expusă. Într-un proces normal de reparare, celulele epiteliale alveolare de tip II mor, iar celulele rămase se răspândesc și suferă un proces de diferențiere pentru a deveni AEC de tip I. În prezența condițiilor patologice și a factorului de creștere beta transformant (TGFβ), fibroblastele se acumulează în aceste zone rănite și se diferențiază în miofibroblaste care secretă colagen și alte proteine. [14] În trecut, se credea că inflamația este principalul factor în cicatrizarea țesutului pulmonar. Cele mai recente rezultate arată însă că dezvoltarea focarelor fibroblastice precede acumularea de celule inflamatorii și, în consecință, depunerea colagenului. [15]
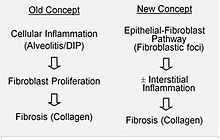
Acest model patogenetic este susținut indirect de caracteristicile clinice ale IPF, inclusiv debutul insidios, progresia pe mai mulți ani, exacerbări acute relativ rare și eșecul de a răspunde la terapia imunosupresoare . [13] [16] În prezent, mai multe terapii care se concentrează pe activarea fibroblastelor sau pe sinteza matricei extracelulare se află în faza inițială de studiu sau sunt luate în considerare pentru dezvoltare.
IPF familial reprezintă mai puțin de 5% din toate cazurile de IPF și nu poate fi distins clinic și histologic de IPF sporadic. [1] Asocierile genetice includ mutații în proteinele tensioactive pulmonare A1, A2, C (SFTPA1, SFTPA2B) și mucină (MUC5B). [17] O caracteristică importantă a variantei MUC5B este frecvența ridicată a detectării acesteia, care este de aproximativ 20% la subiecții din Europa de Nord și de Vest și 19% la populația Framingham Heart Study. [18] Mutațiile genelor telomerazei umane ( telomerază revers transcriptază sau TERT, componenta ARN telomerază sau TERC) sunt, de asemenea, asociate cu fibroza pulmonară familială și, în cazul unora dintre pacienți, cu FPI sporadice. [17] O mutație legată de X într-una din subunitățile telomerazei, diskerina (DKC1) a fost recent descrisă într-o familie cu IPF. [19]
Diagnostic
Diagnosticul precoce al FPI este condiția prealabilă pentru un tratament prompt care poate duce la un rezultat clinic mai bun pe termen lung al acestei boli progresive și fatale. [1] Dacă se suspectează IPF, diagnosticul poate fi dificil; cu toate acestea, sa demonstrat că o abordare multidisciplinară care implică pneumologi, radiologi și patologi cu experiență în boli pulmonare interstițiale contribuie la precizia diagnosticului de FPI. [1] [20] [21] Un document de consens multidisciplinar privind pneumonia interstițială idiopatică a fost publicat de Societatea Americană Toracică (ATS) și Societatea Respiratorie Europeană (ERS) în 2000, propunând criterii specifice majore și minore pentru determinarea diagnosticului IPF. . [1] În 2011, cu toate acestea, au fost publicate noi criterii simplificate și actualizate pentru diagnosticarea și gestionarea IPF de către ATS și ERS, în cooperare cu Societatea Respiratorie Japoneză (JRS) și Asociația Latino-Americană Toracică (ALAT). [1] În prezent, factorii care determină un diagnostic de IPF sunt:
- excluderea cauzelor cunoscute ale ILD, cum ar fi expunerea la domiciliu și mediul profesional, tulburări ale țesutului conjunctiv sau expunerea / toxicitatea la medicamente;
- prezența unui model radiologic tipic UIP la examenul HRCT.
Într-un cadru clinic adecvat, diagnosticul IPF poate fi pus prin utilizarea numai a HRCT, evitând necesitatea unei biopsii pulmonare chirurgicale. [1] [2]
Recunoașterea IPF în practica clinică poate fi dificilă, deoarece simptomele care apar sunt similare cu cele ale bolilor mai frecvente, cum ar fi astmul bronșic ,boala pulmonară obstructivă cronică (BPOC) și insuficiența cardiacă congestivă ( www.diagnoseipf.com ). Principala provocare pentru clinicieni este de a determina dacă istoricul, simptomele (sau semnele ), radiologia și testele funcției pulmonare sunt în concordanță cu un diagnostic de FPI sau dacă rezultatele sunt determinate de un alt proces de boală. De mult timp a fost dificil să se distingă pacienții cu forme de ILD asociate cu expunerea la azbest , medicamente (cum ar fi agenții de chimioterapie sau nitrofurantoina ), artrita reumatoidă și sclerodermia / scleroza sistemică de pacienții cu FPI. Alte considerații pentru diagnosticul diferențial includ boala pulmonară interstițială legată de boala mixtă a țesutului conjunctiv, sarcoidoza avansată, pneumonita de hipersensibilitate cronică, histiocitoza pulmonară a celulelor Langerhans și fibroza radiației. [1] [2]
Caracteristici clinice

La mulți pacienți, simptomele apar cu mult înainte de diagnostic. [3] Cele mai frecvente caracteristici clinice ale IPF includ: [1] [2] [22]
- vârsta peste 50 de ani;
- tuse uscată, neproductivă la efort;
- dispnee progresivă de efort (dificultăți de respirație în timpul exercițiului);
- uscat inspiratorii bibasal raluri , amintind de sunetul velcro, detectate pe auscultation (stetoscopului detectează un sunet în plămâni în timpul inhalării similare cu o deschidere lentă a velcro); [1] [5] [23] hipocratism digital, o deformare a vârfurilor degetelor sau de la picioare (vezi imaginea);
Rezultatele testelor funcției pulmonare nu sunt normale, cu dovezi ale restricției bronșice și ale defectului difuziei alveolare a gazelor.
Aceste caracteristici se datorează lipsei cronice de oxigen din sânge și pot apărea în numeroase alte forme de boli pulmonare, fără a fi specifice IPF. Cu toate acestea, prezența IPF ar trebui luată în considerare la toți pacienții cu dispnee cronică de efort care suferă de tuse, crăpături inspiratorii bibazilare sau hipocratism deget . [1]
Evaluarea prezenței crăpăturilor de tip velcro prin auscultarea plămânilor este o metodă practică pentru a ajuta la diagnosticul precoce al FPI. Crăpăturile subtile sunt ușor recunoscute de medici și sunt caracteristice IPF.[24]
Dacă crackle bilaterale subtile sunt prezente pe întreaga durată a respirației și persistă după mai multe respirații profunde și dacă apar din nou în mai multe ocazii la câteva săptămâni distanță la o persoană cu vârsta ≥ 60 de ani, este recomandabil să luați în considerare ipoteza IPF și , prin urmare , să evalueze executarea unui examen HRCT (rezolutie CT ridicat) a pieptului, mai sensibil decât o toracica radiografie . [23] Deoarece crăpăturile nu sunt specifice IPF, ar trebui inițiat un diagnostic aprofundat. [1]
Radiologie
Pieptului cu raze X este util pentru rutina de urmarire a pacientilor cu IPF. Din păcate, radiografia toracică singură nu este decisivă în scop diagnostic, dar poate identifica totuși o scădere a volumului pulmonar , de obicei cu semne interstițiale reticulare evidente la baza plămânilor. [1]

Evaluarea radiologică prin HRCT este un punct focal în calea diagnosticului IPF. HRCT se efectuează folosind un tomograf axial computerizat obișnuit fără a injecta mediu de contrast. Secțiunile analizei sunt foarte subțiri (1-2 mm).
CT de înaltă rezoluție (HRCT) al pieptului la pacienții cu FPI arată de obicei prezența modificărilor fibrotice la ambii plămâni, cu prevalență la bază și în zonele periferice. Conform orientărilor comune ale ATS / ERS / JRS / ALAT din 2011, HRCT este un element esențial al căii de diagnostic a IPF capabil să identifice formele UIP prin prezența: [1]
- opacități reticulare, adesea asociate cu bronșiectazii de tracțiune;
- plămân fagure de miere caracterizat prin grupuri de spații aeriene chistice, cu un diametru de obicei similar (3-10 mm), ocazional mai mare. În general la nivel sub-pleural și caracterizat prin pereți bine definiți și cu chisturi dispuse în cel puțin două rânduri. De obicei, un singur rând de chisturi nu este suficient pentru a defini aspectul fagurelui;
- opacitatea sticlei mată, frecventă, dar mai puțin extinsă decât modelul reticular;
- distribuție caracteristică la bază și periferic, deși deseori nu este uniformă.

Histologie
Conform orientărilor actualizate din 2011, în absența unui model tipic UIP la examinarea HRCT, trebuie efectuată o biopsie pulmonară pentru un anumit diagnostic. [1] Specimenele histologice pentru diagnosticul IPF trebuie luate cel puțin în trei locații diferite și să fie suficient de mari pentru a permite patologului să evalueze arhitectura pulmonară subiacentă. Fragmente mici de biopsie, cum ar fi cele obținute cu biopsie pulmonară transbronșică (efectuată în timpul bronhoscopiei), nu sunt adesea suficiente în acest scop. Prin urmare, sunt de obicei necesare probe mai mari de biopsie prelevate chirurgical prin toracotomie sau toracoscopie . [1] [2]
Țesutul pulmonar la persoanele cu IPF are de obicei un model histopatologic caracteristic UIP și, prin urmare, este omologul patologic al IPF. [1][4] Deși un diagnostic patologic al UIP corespunde adesea unui diagnostic clinic de IPF, este posibil să se observe un model histologic al UIP și în alte boli și fibroze de origine cunoscută (de exemplu în bolile reumatice). [1] Există patru caracteristici cheie ale UIP-urilor care includ fibroza interstițială "conformație neregulată", leziuni interstițiale, structuri fagure de miere și focare fibroblastice.
Focurile fibroblastice sunt acumulări compacte de miofibroblaste și țesut cicatricial; împreună cu aspectul fagure sunt principalele date patologice care permit diagnosticarea IPF.

Spălarea bronhoalveolară
Spălarea bronhoalveolară (BAL) este o procedură de diagnostic bine tolerată în ILD. [22] Analiza citologiei BAL (număr diferențial de celule) poate fi luată în considerare pentru evaluarea pacienților cu FPI la discreția medicului curant pe baza disponibilității și experienței acestei proceduri în unitatea dumneavoastră. BAL poate duce la diagnostice alternative specifice: malignitate, infecții , pneumonie eozinofilă , histiocitoză X sau proteinoză alveolară. În evaluarea pacienților cu suspiciune de FPI, cea mai importantă aplicație a BAL constă în excluderea altor diagnostice. O creștere izolată a limfocitelor exclude diagnosticul de FPI. [25]
Testul funcției pulmonare
Spirometria identifică de obicei o reducere a capacității vitale (VC) cu o reducere proporțională a ventilației sau o creștere a ventilației pentru capacitatea vitală analizată. Ultima constatare este indicativă a unei rigidități pulmonare mai mari (adaptare pulmonară mai mică) asociată cu fibroza pulmonară, ceea ce duce la o forță de retracție elastică pulmonară mai mare. [26]
Măsurarea statică a volumului pulmonar utilizând pletismografie corporală sau alte tehnici relevă de obicei o reducere a volumului pulmonar (restricție). Acest lucru este reprezentativ pentru dificultățile întâmpinate în expansiunea plămânilor fibrotici.
Capacitatea de difuzie a monoxidului de carbon (DL CO ) este redusă invariabil în IPF și poate fi singura anomalie în boala precoce sau ușoară. Această insuficiență stă la baza înclinației pacienților cu IPF de a prezenta desaturarea oxigenului în timpul efortului, care poate fi, de asemenea, analizată prin utilizarea testului de mers pe jos de 6 minute (6MWT). [1]
Termeni precum „ușor” sau „moderat” și „sever” sunt uneori folosiți pentru clasificarea bolii și se bazează de obicei pe măsurători din testele funcționale pulmonare în repaus. [1] Cu toate acestea, nu există un consens clar cu privire la clasificarea pacienților cu FPI și care sunt cele mai bune criterii și valori de aplicat. IPF ușoară până la moderată este definită de următoarele criterii funcționale: [27] [28] [29] [30]
- capacitate vitală forțată (FVC) ≥50%;
- DL CO ≥30%;
- distanța parcursă în 6MWT ≥150 metri.
Program de consiliere genetică pentru IPF familial
Se estimează că 10-15% dintre pacienții cu FPI au o boală familială, adică recurentă în arborele genetic. Un studiu recent a identificat mutații genetice asociate cu fibroza pulmonară familială (a se vedea mai sus) care pot fi acum detectate datorită testelor de diagnostic dezvoltate și puse la dispoziția publicului. Consilierea genetică oferă informații despre natura, caracterul ereditar și implicațiile unei boli genetice pentru a ajuta indivizii și familiile, atât în alegerile personale, cât și medicale, și pentru a evalua riscul lor de a dezvolta o boală moștenită. Consilierea genetică și screeningul pentru toate mutațiile cunoscute ar trebui făcute mai ales atunci când mai mulți membri ai familiei au fibroză pulmonară. Acest program permite o interpretare personalizată a rezultatelor și a impactului acestora atât asupra sănătății pacientului, cât și asupra altor membri ai familiei.
Prognoză

Cursul clinic al IPF poate fi dificil de prezis. [1] [31] [32] Progresia IPF este asociată cu un timp mediu de supraviețuire estimat la 2-5 ani după diagnostic. [1]
Supraviețuirea la 5 ani de la IPF variază de la 20 la 40%, [32] cu o rată a mortalității mai mare decât multe tipuri de cancer, inclusiv cancer de colon, mielom multiplu și cancer de vezică urinară. [31] [32]
Recent, un indice multidimensional și un sistem de clasificare au fost propuse pentru a prezice mortalitatea în IPF. [33] Denumirea indicelui este GAP (sex, vârstă, fiziologie) și se bazează pe sex, vârstă și două variabile fiziologice pulmonare (FVC și DL CO ) evaluate de obicei în practica clinică pentru a prezice mortalitatea IPF. Valoarea mai mare a indicelui GAP (gradul III) a fost asociată cu un risc de mortalitate de 39% în primul an. [33]
Acest model a fost aplicat și în IPF și în alte ILD, dovedindu-se un bun predictor al mortalității în toate subtipurile majore de ILD. A fost dezvoltat un indice ILD-GAP modificat, aplicabil subtipurilor ILD, care oferă o estimare specifică a supraviețuirii. [34] La pacienții cu FPI, în ciuda ratei ridicate de mortalitate globală pe 5 ani, rata anuală a mortalității pe toate cauzele este relativ scăzută pentru insuficiența ușoară până la moderată a funcției pulmonare. Din acest motiv, studiile clinice de un an cu IPF evaluează schimbarea funcției pulmonare (FVC) mai degrabă decât supraviețuirea. [35] În plus față de parametrii clinici și fiziologici care prezic rata de progresie a IPF, caracteristicile genetice și moleculare sunt corelate cu mortalitatea din IPF. Într-adevăr, s-a demonstrat că pacienții cu un genotip specific din polimorfismul genei mucinei MUC5B prezintă un declin mai lent al FVC și o îmbunătățire semnificativă a supraviețuirii. [36] [37] În ciuda importanței științifice a acestor date, aplicarea unui model predictiv bazat pe evaluarea genotipurilor specifice nu este posibilă în prezent în practica clinică. Pirfenidona , o moleculă cu greutate moleculară mică, este aprobată nu numai în Europa , ci și în Japonia , Coreea de Sud , Canada , China , India , Argentina și Mexic .
Tratament
Obiectivele tratamentului IPF sunt în esență reducerea simptomelor, stoparea progresiei bolii, prevenirea exacerbărilor acute și prelungirea supraviețuirii. Tratamentul preventiv (de exemplu, vaccinurile) și tratamentele simptomatice trebuie începute devreme la fiecare pacient. [38]
Intervenții farmacologice
În trecut au fost studiate numeroase tratamente pentru IPF, inclusiv interferon gamma-1β, [39] bosentan , [40] ambrisentan, [41] și anticoagulante ; [42] cu toate acestea, aceste tratamente nu mai sunt considerate opțiuni terapeutice valabile astăzi. Multe dintre aceste studii inițiale s-au bazat pe ipoteza că IPF a fost o boală inflamatorie.
Pirfenidonă
Pirfenidona este o moleculă mică care combină atât efectele antiinflamatorii, cât și cele anti-oxidante și anti-fibrotice în modele experimentale de fibroză.[43] Pirfenidona, comercializată sub denumirea comercială Esbriet , este aprobată în Europa pentru tratamentul pacienților cu FPI ușoară până la moderată. Este, de asemenea, aprobat în Japonia și Coreea de Sud (denumirea comercială Pirespa), precum și în Canada, China, India, Argentina și Mexic.
Pirfenidona a fost aprobată în Uniunea Europeană pe baza rezultatelor a trei studii de fază III randomizate, dublu-orb, controlate cu placebo: unul efectuat în Japonia, celelalte două în Europa și Statele Unite (studii CAPACITY ). [27] [44]
O revizuire din Biblioteca Cochrane (jurnalul Cochrane Collaboration for evidence-based Medicine) bazată pe patru studii care au implicat 1155 de pacienți care au comparat pirfenidonă cu placebo au arătat o reducere semnificativă cu 30% a progresiei bolii la pacienții tratați cu pirfenidonă. [45] Valorile FVC sau VC au fost, de asemenea, îmbunătățite semnificativ cu pirfenidonă, deși s-a arătat o ușoară scădere a FVC doar în unul dintre cele două studii CAPACITY. [27] Pe baza acestor rezultate contradictorii, Administrația Federală Americană pentru Alimente și Medicamente (FDA) a solicitat un al treilea studiu clinic de fază III, ASCEND , în curs de desfășurare în Statele Unite. Acest studiu, finalizat în 2014 și publicat online în New England Journal of Medicine, a arătat că pirfenidona reduce semnificativ declinul funcției pulmonare și progresia IPF. [29] Rezultatele studiului ASCEND au fost, de asemenea, combinate cu cele din cele două studii CAPACITY într-o analiză prespecificată care a demonstrat că pirfenidona reduce mortalitatea cu aproximativ 50% pe parcursul unui an de tratament. [29] Pe baza acestor rezultate, pirfenidona a primit definiția FDA de Breakthrough Therapy, o definiție rezervată acelor medicamente destinate tratamentului afecțiunilor grave sau fatale ale căror date clinice preliminare au arătat o îmbunătățire substanțială a unuia sau mai multor obiective în raport cu cele existente terapii. [46]
Compania farmaceutică care a dezvoltat pirfenidonă, InterMune Inc., a furnizat inițial medicamentul pentru utilizare cu compasiune în Statele Unite și Europa, deja în pregătirea aprobării de comercializare printr-un program de acces extins (EAP) [47] . Din 2014, medicamentul a fost prescris în mod regulat în Italia de centre specializate identificate la nivelul regiunilor individuale. Medicamentul este comercializat în prezent de Roche după achiziționarea InterMune.
N-acetilcisteina și tripla terapie
N-acetilcisteina (NAC) este un precursor al glutationului, un antioxidant . Se presupune că tratamentul cu doze mari de NAC poate acționa asupra dezechilibrului oxidant-antioxidant care apare în țesutul pulmonar al pacienților cu FPI. În primul studiu clinic care a implicat 180 de pacienți (IFIGENIA), sa demonstrat că terapia NAC reduce scăderea VC și DL CO în 12 luni de urmărire atunci când a fost luată în asociere cu prednison și azatioprină (terapie triplă). [48]
Mai recent, un mare studiu randomizat controlat (PANTHER-IPF) a fost inițiat de National Institutes of Health (NIH) din Statele Unite pentru a evalua terapia triplă și monoterapia cu NAC la pacienții cu IPF. Questo studio ha rilevato che la combinazione di prednisone, azatioprina e NAC aumenta il rischio di morte e il numero di ospedalizzazioni; il NIH ha quindi annunciato nel 2012 la chiusura anticipata del braccio in tripla terapia dello studio PANTHER-IPF. [49] Lo studio ha concluso che “acetilcisteina rispetto a placebo, non ha offerto alcun beneficio significativo per quanto riguarda il mantenimento della FVC in pazienti con fibrosi polmonare idiopatica con compromissione della funzionalità polmonare da lieve a moderata". [50]
Questo studio ha anche valutato il solo trattamento con N-acetilcisteina (NAC). I dati ottenuti, recentemente pubblicati su New England Journal of Medicine, hanno dimostrato che la monoterapia con NAC non comporta benefici significativi nei pazienti con IPF da lieve a moderata. [51]
Nintedanib (precedentemente BIBF 1120)
Una molecola in fase di sviluppo ha completato due studi clinici di Fase III (INPULSIS-1 e INPULSIS-2). [30] Nintedanib è un triplice inibitore sperimentale dell'angiochinasi che ha come bersaglio i recettori tirosin-chinasici coinvolti nella regolazione dell'angiogenesi e nella patogenesi della fibrosi e della IPF: il recettore del fattore di crescita dei fibroblasti (FGFR), il recettore del fattore di crescita derivato dalle piastrine (PDGFR) e il recettore del fattore di crescita dell'endotelio vascolare (VEGFR). [52] In entrambi gli studi di Fase III, nintedanib ha significativamente ridotto il declino della funzionalità polmonare di circa il 50% in un anno di trattamento. [30] Per quanto riguarda il raggiungimento degli endpoint secondari, solo nello studio INPULSIS-2 si è verificato un significativo aumento del tempo (ritardo) alla prima riacutizzazione grave nel gruppo in trattamento con nintedanib rispetto a placebo. Questo incremento non si è verificato nello studio INPULSIS-1. Nintedanib, come pirfenidone, ha ricevuto l'approvazione alla compilazione del file FDA come Priority Review. [53]
Opzioni terapeutiche in corso di sperimentazione
Le molecole attualmente in sperimentazione per la IPF in studi clinici di Fase II includono gli anticorpi monoclonali simtuzumab, tralokimab, lebrikizumab e FG-3019 e l'antagonista del recettore dell'acido lisofosfatidico BMS-986020. È in corso, inoltre, uno studio di Fase II per la molecola STX-100. [54] Queste molecole hanno come bersaglio diversi fattori di crescita e citochine che rivestono un ruolo nella proliferazione, nell'attivazione, nella differenziazione o nell'alterazione della sopravvivenza dei fibroblasti.
È possibile reperire ulteriori informazioni alla pagina www.ClinicalTrials.gov, un registro e banca dati di risultati di studi clinici con finanziamento pubblico e privato a partecipazione umana svolti nel mondo.
Interventi non farmacologici
Trapianto di polmone
Il trapianto polmonare può essere utile nei pazienti candidabili da un punto di vista fisico a essere sottoposti a un intervento di tale portata. È stato dimostrato che nei pazienti affetti da IPF il trapianto di polmone riduca il rischio di decesso del 75% rispetto ai pazienti che restano in lista d'attesa. [55] Dall'introduzione del punteggio di allocazione del polmone (LAS), che classifica i candidati al trapianto sulla base della probabilità di sopravvivenza, la IPF è divenuta l'indicazione più frequente per il trapianto di polmone negli Stati Uniti. [56]
I pazienti sintomatici affetti da IPF con età inferiore ai 65 anni e con un indice di massa corporea (BMI) ≤26 kg/m2 dovrebbero essere candidati al trapianto polmonare; non esistono tuttavia dei dati chiari che indichino il momento preciso per il trapianto. Sebbene controversi, i dati più recenti indicano che il trapianto bilaterale polmonare è prevalente rispetto al trapianto polmonare singolo nei pazienti con IPF. [57] Il tasso di sopravvivenza a cinque anni dal trapianto di polmone è stimato tra 50% e 56%. [1] [58] [59]
Ossigenoterapia a lungo termine (LTOT)
Nelle linee guida sulla IPF del 2011, l' ossigenoterapia , od ossigenazione integrativa per uso domestico, è divenuta una raccomandazione per i pazienti con ipossiemia a riposo clinicamente significativa. Sebbene non sia stato dimostrato che l'ossigenoterapia migliori la sopravvivenza nei pazienti con IPF, alcuni dati indicano un miglioramento nella capacità di svolgere esercizio fisico. [1] [60]
Riabilitazione polmonare
La stanchezza e la perdita di massa muscolare sono problematiche frequenti e debilitanti per i pazienti con IPF. La riabilitazione polmonare può alleviare i sintomi manifesti di IPF e migliorare la funzionalità stabilizzando e/o invertendo le caratteristiche extrapolmonari della malattia. [38] [56] Il numero di studi pubblicati sul ruolo della riabilitazione polmonare nella fibrosi polmonare idiopatica è ridotto, tuttavia nella maggior parte di questi studi sono stati riscontrati miglioramenti significativi a breve termine sulla tolleranza all'esercizio, sulla qualità di vita e sulla dispnea da sforzo. [61] I principali programmi di riabilitazione prevedono esercizio fisico, regolazione dell'alimentazione, terapia occupazionale , supporto informativo, e terapia psicosociale.
Nella fase avanzata della malattia, i pazienti con IPF tendono a interrompere l'attività fisica a causa dell'aumento di dispnea. Dove possibile, è opportuno scoraggiare l'interruzione dell'attività fisica.
Cure palliative
Le cure palliative si concentrano soprattutto sulla riduzione dei sintomi e sul miglioramento della condizione del paziente, più che sul trattamento della malattia. Vi può essere il trattamento di sintomi in fase di peggioramento con l'utilizzo di oppioidi cronici per dispnea acuta e tosse. Anche l'ossigenoterapia può essere utile come cura palliativa della dispnea nei pazienti ipossiemici.
Tra le cure palliative vi sono anche il supporto fisico ed emotivo e il sostegno psicosociale per i pazienti ei loro familiari. [1] Con la progressione della malattia, i pazienti possono essere spaventati e andare incontro ad ansia e depressione: è pertanto consigliato prendere in considerazione un supporto psicologico. Uno studio recente su pazienti ambulatoriali con ILD, tra cui la IPF, ha mostrato come il grado di depressione, la funzionalità generale (sulla base dei risultati del test di cammino) e la funzionalità polmonare contribuiscano alla gravità della dispnea. [62]
È possibile prendere in considerazione l'uso di morfina in casi specifici di dispnea particolarmente acuta. La morfina può ridurre la dispnea, l'ansia e la tosse senza andare a ridurre in modo significativo la saturazione di ossigeno. [63]
Gestione e follow-up
Sono frequenti i casi di diagnosi errata di IPF, perlomeno fino a che i dati fisiologici e/o di imaging non indichino la presenza di una ILD, determinando un ritardo nell'avvio delle cure appropriate. [56] Considerando che la IPF è una patologia con una sopravvivenza media di 3 anni dalla diagnosi, sarebbe opportuno indirizzare tempestivamente qualsiasi paziente con ILD manifesta o sospetta a un centro con competenze specifiche. Sulla base della complessità della diagnosi differenziale, è estremamente importante, ai fini di una diagnosi accurata, uno scambio multidisciplinare tra pneumologo, radiologo e patologo esperti nella diagnosi di ILD. [1]
A seguito della diagnosi di IPF e di un'adeguata scelta terapeutica sulla base dei sintomi e dello stadio della malattia, è opportuno effettuare uno stretto follow-up del paziente. A causa del decorso estremamente variabile della malattia e dell'elevata incidenza di complicanze, quali il cancro al polmone (fino al 25% dei pazienti con IPF), è indispensabile effettuare delle analisi di routine ogni 3-6 mesi, che includano la spirometria (pletismografia corporea), il test della capacità di diffusione, il 6MWT, oltre alla valutazione della dispnea, della qualità della vita e della necessità di ossigeno.
Inoltre, la maggiore consapevolezza delle complicanze e delle condizioni cliniche concomitanti spesso associate alla IPF, richiede una valutazione di routine delle comorbidità, la maggior parte delle quali riflette semplicemente malattie concomitanti di invecchiamento, l'uso di farmaci e la loro interazione e gli effetti collaterali.
Esacerbazioni acute
Le esacerbazioni acute della IPF (AE-IPF) vengono definite come un peggioramento immotivato o lo sviluppo di dispnea nell'arco di 30 giorni con la presenza di nuovi infiltrati radiologici all'esame HRCT, spesso sovrapposti a un pattern di fondo compatibile con UIP. L'incidenza precoce delle AE-IPF varia dal 10% al 15% di tutti i pazienti. La prognosi di AE-IPF è infausta, con un tasso di mortalità compreso tra il 78% e il 96%. [64] Vanno escluse altre cause di AE-IPF, quali l'embolia polmonare, lo scompenso cardiaco congestizio, lo pneumotorace o l'infezione. L'infezione polmonare deve essere esclusa tramite l'aspirato endotracheale o il BAL.
Molti pazienti che subiscono un peggioramento acuto necessitano di trattamenti intensivi, soprattutto se lo scompenso respiratorio si associa a instabilità emodinamica, comorbidità importanti o ipossiemia acuta. [65] Tuttavia, il tasso di mortalità durante l'ospedalizzazione è elevato. [64] La ventilazione meccanica va introdotta solamente dopo un'attenta valutazione della prognosi a lungo termine del paziente e, dove possibile, tenendo in considerazione le sue volontà. Le attuali linee guida, tuttavia, scoraggiano l'utilizzo della ventilazione meccanica nei pazienti con scompenso respiratorio conseguente a IPF. [1]
In altre specie
La IPF è stata individuata in diverse razze di cani e di gatti;[66] la sua manifestazione più tipica si riscontra nelWest Highland White Terrier . [67] Gli animali che ne sono affetti, condividono molte delle manifestazioni cliniche dell'uomo, tra cui una maggiore e progressiva intolleranza all'esercizio fisico, una frequenza respiratoria più elevata e una possibile disfunzione respiratoria. [68] La prognosi solitamente è infausta. [ non chiaro ]
Note
- ^ a b c d e f g h i j k l m n o p q r s t u v w x y z aa ab ac ad ae af ag ah ai aj ak Raghu G, Collard HR, Egan JJ, et al., An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management , in Am J Respir Crit Care Med , vol. 183, n. 6, 2011, pp. 788–824, DOI : 10.1164/rccm.2009-040GL , PMID 21471066 .
- ^ a b c d e f g h i j k l m n American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias, This official statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was approved by the ATS board of directors, June 2013 and by the ERS Steering Committee, March 2013 , in Am Respir Crit Care Med , vol. 188, n. 6, 2013, pp. 733-748, PMID 24032382 .
- ^ a b Meltzer EB, Noble PW,Idiopathic pulmonary fibrosis , in Orphanet J Rare Dis , vol. 3, n. 1, 2008, DOI : 10.1186/1750-1172-3-8 , PMC 2330030 , PMID 18366757 .
- ^ a b c d e Pulmonary Fibrosis Foundation. “Prevalence and Incidence”. Pulmonaryfibrosis.org. Retrieved 2013-04-11
- ^ a b c Raghu G, Weycker D, Edesberg J, Bradford WZ, Oster G, Incidence and prevalence of idiopathic pulmonary fibrosis , in Am. J Respir. Crit. Care Med , vol. 174, n. 7, 2006, pp. 810–816.
- ^ Gribbin J, Hubbard RB, Le Jeune I, Smith CJ, West J, Tata LJ, Incidence and mortality of idiopathic pulmonary fibrosis and sarcoidosis in the UK , in Thorax , vol. 61, n. 11, 2006, pp. 980-985, PMC 2121155 , PMID 16844727 .
- ^ Eurostat News Release. European demography. 110/2010. 27 July 2010
- ^ Hyldgaard C, Hilberg O, Muller A, Bendstrup E, A cohort study of interstitial lung diseases in central Denmark , in Respir Med , vol. 108, n. 5, 2014, pp. 793–799, PMID 24636811 .
- ^ a b Olson AL, Swigris JJ, Idiopathic pulmonary fibrosis: diagnosis and epidemiology , in Clinics in chest medicine , vol. 33, n. 1, 2012, pp. 41–50, DOI : 10.1016/j.ccm.2011.12.001 , PMID 2236524 .
- ^ a b Williams KJ, Gammaherpesviruses and Pulmonary Fibrosis: Evidence From Humans, Horses, and Rodents , in Veterinary Pathology , vol. 51, n. 2, 2014, pp. 372–384, DOI : 10.1177/0300985814521838 , PMID 24569614 .
- ^ García-Sancho C, Buendía-Roldán I, Fernández-Plata MR, Navarro C, Pérez-Padilla R, Vargas MH, Loyd JE, Selman M. Buendía-Roldán I, Fernández-Plata MR, et al., Familial pulmonary fibrosis is the strongest risk factor for idiopathic pulmonary fibrosis , in Respir Med. , vol. 105, n. 12, 2011, pp. 1902–1990, PMID 21917441 .
- ^ a b Harari S, Caminati A, IPF: new insight on pathogenesis and treatment , in Allergy , vol. 65, n. 5, 2010, pp. 537–553, DOI : 10.1111/j.1398-9995.2009.02305 , PMID 20121758 .
- ^ a b Loomis-King H, Flaherty KR, Moore BB, Pathogenesis, current treatments and future directions for idiopathic pulmonary fibrosis , in Curr. Opin. Pharmacol. , vol. 13, n. 3, aprile 2013, pp. 377-385, DOI : 10.1016/j.coph.2013.03.015 .
- ^ Pardo A, Selman M, Idiopathic pulmonary fibrosis: new insights in its pathogenesis , in Int J Biochem Cell Biol. , vol. 34, n. 12, 2002, pp. 1534–1538.
- ^ Selman M, King TE, Pardo A, Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy , in Ann Intern Med. , vol. 134, n. 2, 2001, pp. 136–151, PMID 11177318 .
- ^ a b Online 'Mendelian Inheritance in Man' (OMIM) 178500
- ^ Mathai S, et al., Genetic susceptibility and pulmonary fibrosis , in Curr Opin Pulm Med , vol. 20, n. 5, 2014, pp. 429–435, PMID 25022318 .
- ^ Kropski JA, Mitchell DB, Markin C, et al., A novel dyskerin (DKC1) mutation is associated with familial interstitial pneumonia , in Chest , 2014, PMID 24504062 .
- ^ Flaherty KR, King TE, Raghu G, Lynch JP, Colby TV, Travis WD, Gross BH, Kazerooni EA, et al., Idiopathic interstitial pneumonia: what is the effect of a multidisciplinary approach to diagnosis? , in Am J Respir Crit Care Med. , vol. 170, n. 8, 2004, pp. 904-910, DOI : 10.1164/rccm.200402-147OC , PMID 15256390 .
- ^ Flaherty KR, Andrei AC, King TE Jr, Raghu G, Colby TV, Wells A, Bassily N, Brown K, et al.,Idiopathic interstitial pneumonia: do community and academic physicians agree on diagnosis? , in Am J Respir Crit Care Med. , vol. 175, n. 10, 2007, pp. 1054–1060, PMC 1899268 , PMID 17255566 .
- ^ a b Flaherty KR, Mumford JA, Murray S, Kazerooni EA, Gross BH, Colby TV, Travis WD, Flint A, et al., Prognostic implications of physiologic and radiographic changes in idiopathic interstitial pneumonia , in Am J Respir Crit Care Med. , vol. 168, n. 5, 2007, pp. 543-548, PMID 12773329 .
- ^ a b Cottin V, Cordier JF, Velcro crackles: the key for early diagnosis of idiopathic pulmonary fibrosis , in Eur Respir J. , vol. 40, n. 3, 2012, pp. 519-521, DOI : 10.1183/09031936.00001612 , PMID 22941541 .
- ^ Baughman RP, Shipley RT, Loudon RG, Lower EE, Crackles in interstitial lung disease. Comparison of sarcoidosis and fibrosing alveolitis , in Chest , vol. 100, n. 1, 1991, pp. 96-101, PMID 2060395 .
- ^ Ohshimo S, Bonella F, Cui A, Beume M, Kohno N, Guzman J, Costabel U, Significance of bronchoalveolar lavage for the diagnosis of idiopathic pulmonary fibrosis , in Am J Respir Crit Care Med. , vol. 179, n. 11, 2009, pp. 1043–1047.
- ^ Pellegrino R, Viegi G, Brusasco V, Crapo RO, Burgos F, Casaburi R, Coates A, van der Grinten CP, et al., Interpretative strategies for lung function tests , in Eur Respir J. , vol. 26, n. 5, 2005, pp. 948–968, DOI : 10.1183/09031936.05.00035205 , PMID 16264058 .
- ^ a b c Noble PW, Albera C, Bradford WZ, Costabel U, Glassberg MK, Kardatzke D, King TE, Lancaster L, et al., Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials , in Lancet , vol. 377, n. 9779, 2011, pp. 1760–1769, DOI : 10.1016/S0140-6736(11)60405-4 , PMID 21571362 .
- ^ Martinez FJ, Safrin S, Weycker D, Starko KM, Bradford WZ, King TE Jr, Flaherty KR, Schwartz DA, et al. IPF study group, The clinical course of patients with idiopathic pulmonary fibrosis , in Ann Intern Med. , vol. 377, n. 9779, 2005, pp. 1760–1769, DOI : 10.1016/S0140-6736(11)60405-4 , PMID 21571362 .
- ^ a b c King TE Jr, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, Gorina E, Hopkins PM, for the ASCEND Study Group., A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis , in N Engl J Med , vol. 370, n. 22, 2014, pp. 2083–2092, PMID 24836312 .
- ^ a b c Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, M Costabel U, Cottin V, Flaherty KR, for the INPULSIS Trial Investigators., Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis , in N Engl J Med , vol. 370, n. 22, 2014, pp. 2071–2082, PMID 24836310 .
- ^ a b c Bjoraker JA, Ryu JH, Edwin MK, Myers JL, Tazelaar HD, Schroeder DR, Offord KP, Prognostic significance of histopathologic subsets in idiopathic pulmonary fibrosis , in Am J Respir Crit Care Med. , vol. 157, n. 1, 1998, pp. 199-203, PMID 9445300 .
- ^ a b c Kim DS, Collard HR, King TE,Classification and natural history of the idiopathic interstitial pneumonias , in Proc Am Thorac Soc. , vol. 3, n. 4, 2006, pp. 285-292, DOI : 10.1513/pats.200601-005TK , PMC 2658683 , PMID 1673819 .
- ^ a b Ley B, Ryerson CJ, Vittinghoff E, Ryu JH, Tomassetti S, Lee JS, Poletti V, Buccioli M, Elicker BM, Jones KD, King TE Jr, Collard HR, A multidimensional index and staging system for idiopathic pulmonary fibrosis , in Ann Intern Med. , vol. 156, n. 10, pp. 684–691.
- ^ Ryerson CJ, Vittinghoff E, Ley B, Lee JS, Mooney JJ, Jones KD, Elicker BM, Wolters PJ, et al., Predicting Survival Across Chronic Interstitial Lung Disease: The ILD-GAP Model , in Chest , vol. 145, n. 4, 2014, pp. 723–728, PMID 24114524 .
- ^ King TE Jr, Albera C, Bradford WZ, Costabel U, du Bois RM, Leff JA, Nathan SD, Sahn SA, et al., All-cause mortality rate in patients with idiopathic pulmonary fibrosis. Implications for the design and execution of clinical trials , in Am J Respir Crit Care Med , vol. 189, n. 7, pp. 825–831.
- ^ Peljto AL, Zhang Y, Fingerlin TE, Ma SF, Garcia JG, Richards TJ, Silveira LJ, Lindell KO, et al., Association between the MUC5B promoter polymorphism and survival in patients with idiopathic pulmonary fibrosis , in JAMA , vol. 309, n. 21, 2013, pp. 2232–2239, PMID 23695349 .
- ^ Stock CJ, Sato H, Fonseca C, Banya WA, Molyneaux PL, Adamali H, Russell AM, Denton CP, et al., Mucin 5B promoter polymorphism is associated with idiopathic pulmonary fibrosis but not with development of lung fibrosis in systemic sclerosis or sarcoidosis , in Thorax , vol. 68, n. 5, 2013, pp. 436–441, PMID 23321605 .
- ^ a b Lee JS, McLaughlin S, Collard HR, Comprehensive care of the patient with idiopathic pulmonary fibrosis , in Curr Opin Pulm Med. , vol. 17, n. 5, 2011, pp. 348-354.
- ^ King TE Jr, Albera C, Bradford WZ, Costabel U, Hormel P, Lancaster L, Noble PW, Sahn SA, et al. INSPIRE Study Group, Effect of interferon gamma-1b on survival in patients with idiopathic pulmonary fibrosis , in Lancet , vol. 374, n. 9685, 2009, pp. 222-228, DOI : 10.1016/S0140-6736(09)60551-1 , PMID 19570573 .
- ^ King TE Jr, Brown KK, Raghu G, du Bois RM, Lynch DA, Martinez F, Valeyre D, Leconte I, et al., BUILD-3: a randomized, controlled trial of bosentan in idiopathic pulmonary fibrosis , in Am J Respir Crit Care Med. , vol. 184, n. 1, 2011, pp. 92-99, DOI : 10.1164/rccm.201011-1874OC , PMID 21474646 .
- ^ Raghu G, Behr J, Brown KK, Egan JJ, Kawut SM, Flaherty KR, Martinez FJ, Nathan SD, et al., Treatment of idiopathic pulmonary fibrosis with ambrisentan: a parallel, randomized trial , in Ann Intern Med. , vol. 158, n. 9, 2013, pp. 641-649, DOI : 10.7326/0003-4819-158-9-201305070-00003 , PMID 23648946 .
- ^ Noth I, Anstrom KJ, Calvert SB, de Andrade J, Flaherty KR, Glazer C, Kaner RJ, Olman MA, Idiopathic Pulmonary Fibrosis Clinical Research Network (IPFnet) A placebo-controlled randomized trial of warfarin in idiopathic pulmonary fibrosis , in Am J Respir Crit Care Med. , vol. 186, n. 1, 2012, pp. 88-95, DOI : 10.1164/rccm.201202-0314OC , PMID 22561965 .
- ^ Schaefer CJ, Ruhrmund DW, Pan L, Seiwert SD, Kossen K, Antifibrotic activities of pirfenidone in animal models , in Eur Respir Rev. , vol. 20, n. 120, 2011, pp. 85-97.
- ^ Taniguchi H, Ebina M, Kondoh Y, Ogura T, Azuma A, Suga M, Taguchi Y, Takahashi H, et al., Pirfenidone in idiopathic pulmonary fibrosis , in Eur Respir J. , vol. 35, n. 4, 2010, pp. 821-829, DOI : 10.1183/09031936.00005209 , PMID 19996196 .
- ^ Spagnolo P, Del Giovane C, Luppi F, Cerri S, Balduzzi S, Walters EH, D'Amico R, Richeldi L, Non-steroid agents for idiopathic pulmonary fibrosis , in Cochrane Database Syst Rev , n. 9, 2010, pp. 821-829, DOI : 10.1002/14651858.CD003134.pub2 .
- ^ InterMune Receives FDA Breakthrough Therapy Designation for Pirfenidone, an Investigational Treatment for IPF, Press release. Retrieved 2014-04-08: http://investor.intermune.com/phoenix.zhtml?c=100067&p=irol-newsArticle&ID=1948523&highlight= .
- ^ InterMune Announces Expanded Access Program for Pirfenidone to Treat Idiopathic Pulmonary Fibrosis (IPF) in the United States., Press release. Retrieved 2014-04-08: http://investor.intermune.com/phoenix.zhtml?c=100067&p=irol-newsArticle&ID=1931863&highlight= .
- ^ Demedts M, Behr J, Buhl R, Costabel U, Dekhuijzen R, Jansen HM, MacNee W, Thomeer M, et al., High-dose acetylcysteine in idiopathic pulmonary fibrosis , in N Engl J Med. , vol. 353, n. 21, 2005, pp. 2229–2242, DOI : 10.1056/NEJMoa042976 , PMID 16306520 .
- ^ Commonly used three-drug regimen for idiopathic pulmonary fibrosis found harmful [1] , Nih.gov., Retrieved 2013-04-11
- ^ Behr J., Prednisone, azathioprine an N-acetylcysteine for pulmonary fibrosis , in N Engl J Med. , vol. 367, n. 9, 2012, pp. 869–871, DOI : 10.1056/NEJMc1207471 , PMID 22931324 .
- ^ The Idiopathic Pulmonary Fibrosis Clinical Research Network, Randomized trial of acetylcysteine in idiopathic pulmonary fibrosis , in N Engl J Med. , vol. 370, n. 22, 2014, pp. 2093–2102, PMID 24836309 .
- ^ BIBF 1120 Fact Sheet. Retrieved 2014-04-08
- ^ Boehringer Ingelheim's Investigational Therapy Nintedanib Receives FDA Breakthrough Therapy Designation. Press release. Retrieved 2014-04-08: [2]
- ^ [3] [ collegamento interrotto ]
- ^ Russo MJ, Iribarne A, Hong KN, Davies RR, Xydas S, Takayama H, Ibrahimiye A, Gelijns AC, Bacchetta MD, D'Ovidio F, Arcasoy S, Sonett JR, High lung allocation score is associated with increased morbidity and mortality following transplantation , in Chest , vol. 137, n. 3, 2010, pp. 651-657.
- ^ a b c Spagnolo P, Tonelli R, Cocconcelli E, Stefani A, Richeldi L, Idiopathic pulmonary fibrosis: diagnostic pitfalls and therapeutic challenges , in Multidiscip Respir Med. , vol. 7, n. 1, 2012, p. 42.
- ^ George TJ, Arnaoutakis GJ, Shah AS, Lung transplantation for idiopathic pulmonary fibrosis , in Ann Thorac Surg. , vol. 84, n. 4, 2007, pp. 1121–1128, PMID 17888957 .
- ^ Mason DP, Brizzio ME, Alster JM, McNeill AM, Murthy SC, Budev MM, Mehta AC, Minai OA, et al., Lung transplant in idiopathic pulmonary fibrosis , in Arch Surg. , vol. 146, n. 10, 2011, pp. 1204–1209.
- ^ Keating D, Levvey B, Kotsimbos T, Whitford H, Westall G, Williams T, Snell G, Lung transplantation in pulmonary fibrosis challenging early outcomes counter balanced by surprisingly good outcomes beyond 15 years , in Transplant Proc. , vol. 41, n. 1, 2009, pp. 289–291, DOI : 10.1016/j.transproceed.2008.10.042. , PMID 19249537 .
- ^ Morrison DA, Stovall JR., Increased exercise capacity in hypoxemic patients after long-term oxygen therapy , in Chest , vol. 102, n. 2, 1992, pp. 542-550, PMID 1643945 .
- ^ Kenn K, Gloeckl R, Behr J, Pulmonary rehabilitation in patients with idiopathic pulmonary fibrosis--a review , in Respiration; international review of thoracic diseases , vol. 86, n. 2, 2013, pp. 89–99, DOI : 10.1159/000354112 , PMID 23942353 .
- ^ Ryerson CJ, Berkeley J, Carrieri-Kohlman VL, Pantilat SZ, Landefeld CS, Collard HR, Depression and functional status are strongly associated with dyspnea in interstitial lung disease , in Chest , vol. 139, n. 3, 2011, pp. 609-616.
- ^ Allen S, Raut S, Woollard J, Vassallo M, Low dose diamorphine reduces breathlessness without causing a fall in oxygen saturation in elderly patients with end-stage idiopathic pulmonary fibrosis , in Palliat Med. , vol. 19, n. 2, 2005, pp. 128–130.
- ^ a b Agarwal R, Jindal SK, Acute exacerbation of idiopathic pulmonary fibrosis: a systematic review , in Eur J Intern Med. , vol. 19, n. 4, 2008, pp. 227–235.
- ^ Stern JB, Mal H, Groussard O, Brugière O, Marceau A, Jebrak G, Fournier M, Prognosis of patients with advanced idiopathic pulmonary fibrosis requiring mechanical ventilation for acute respiratory failure , in Chest , vol. 120, n. 1, 2001, pp. 213-219.
- ^ Williams K, Malarkey D, Cohn L, Patrick D, Dye J, Toews G, Identification of spontaneous feline idiopathic pulmonary fibrosis: morphology and ultrastructural evidence for a type II pneumocyte defect , in Chest , vol. 125, n. 6, 2004, pp. 2278–2288, PMID 15189952 .
- ^ Webb JA, Armstrong J, Chronic idiopathic pulmonary fibrosis in a West Highland white terrier , in Can Vet J. , vol. 43, n. 9, 2002, pp. 703–705, PMC 339552 , PMID 12240528 .
- ^ Canine Pulmonary Fibrosis. [4] Akcchf.org. Retrieved 2013-04-11.
Altri progetti
-
 Wikimedia Commons contiene immagini o altri file sulla fibrosi polmonare idiopatica
Wikimedia Commons contiene immagini o altri file sulla fibrosi polmonare idiopatica
Collegamenti esterni
- Associazione Italiana Malattie Interstiziali o rare del Polmone , su aimip.org . URL consultato il 21 novembre 2007 (archiviato dall' url originale il 13 novembre 2007) .
- Comunità Fibrosi Polmonare Idiopatica (IPF) , su rareconnect.org .
- Pulmonary fibrosis foundation , su pulmonaryfibrosis.org . URL consultato il 29 agosto 2013 (archiviato dall' url originale il 3 settembre 2013) .
- Registro europeo sulla IPF , su pulmonary-fibrosis.net .
- ILD CARE FOUNDATION , su ildcare.nl .
- Pulmonary fibrosis foundation , su pulmonaryfibrosis.org . URL consultato il 29 agosto 2013 (archiviato dall' url originale il 3 settembre 2013) .
- IPF - British Lung Foundation , su blf.org.uk .
- The European IPF Registry (eurIPFreg) has become Europe's leading database of longitudinal data from IPF patients, including control groups of patients with other lung diseases , su pulmonary-fibrosis.net .
- Coalition for Pulmonary Fibrosis , su coalitionforpf.org . URL consultato il 26 settembre 2014 (archiviato dall' url originale il 6 novembre 2014) .
- ILD CARE FOUNDATION´s activity is focused to increase knowledge, support research, contribute to prevention and provide counselling for interstitial lung diseases , su ildcare.nl .
- www.diagnoseipf.com , su diagnoseipf.com . URL consultato il 26 settembre 2014 (archiviato dall' url originale il 4 settembre 2014) .
- KnowIPFNow.com .
- inIPF , su inipf.com .
- IPFtoday.com .
- ipfcharter.org . URL consultato il 2 agosto 2020 (archiviato dall' url originale il 22 ottobre 2014) .
- Federazione Italiana IPF e Malattie Rare Polmonari - FIMARP onlus