Sindromul Saethre-Chotzen
| Sindromul Saethre-Chotzen | |
|---|---|
 | |
| Boala rara | |
| Specialitate | reumatologie |
| Clasificare și resurse externe (EN) | |
| ICD-9 -CM | xxx |
| ICD-10 | Q87.0 |
| OMIM | 101400 |
| Sinonime | |
| Tipul 3 acrocefalosindactilie Acrocefalie, asimetrie craniană și sindactilie ... | |
| Eponime | |
| Haakon Saethre F. Chotzen ... | |
Sindromul Saethre-Chotzen , cunoscut și sub numele de acrocefalosindactilie de tip 3 , este o boală genetică rară caracterizată prin craniosinostoză (închiderea prematură a suturilor prezente între oasele craniului ) care provoacă o formă anormală a feței și a craniului: capul suferinței din sindrom tinde spre o formă conică, cu o asimetrie facială mai mult sau mai puțin severă. Boala implică, de asemenea, ptoza pleoapelor , hipertelorismul ocular și sindactilia mâinilor și picioarelor. [1] Persoanele cu o formă deosebit de severă a sindromului au o întârziere mentală ușoară sau moderată. În funcție de amploarea anomaliilor cauzate de sindrom, persoanele afectate pot necesita tratament chirurgical . [2] Majoritatea celor care suferă, totuși, pot trăi o viață relativ normală. [1]
Epidemiologie și note istorice
În 1931 Haakon Saethre, un psihiatru norvegian, a descris caracteristici fenotipice similare între o mamă și cele două fiice ale ei: toate trei aveau o față alungită cu trăsături somatice anormale, degete scurte și sindactilie bilaterală între index și mijloc, precum și între a doua, a treia și a doua al patrulea deget de la picior al fiecărui picior. Un an mai târziu, F. Chotzen, un psihiatru german, a descris o altă familie (un tată și cei doi copii ai săi) cu caracteristici similare cu cele ale celor trei persoane descrise de Saethre, dar cu prezența, de asemenea, a unei întârzieri mintale ușoare, a pierderii auzului și a scurtei statură. Din unirea observațiilor lor, inițial prezentate individual, a derivat numele (Saethre-Chotzen de fapt) al sindromului.
Saethre-Chotzen este cea mai frecventă cauză sindromică a craniosinostozei, deoarece are o incidență estimată a unui caz la fiecare 25.000-50.000 de nașteri vii. [3] Nu are predilecții de gen sau rasă . Dacă un părinte are o copie mutată a genei, are 50% șanse să o transmită copilului său, care ar manifesta în consecință sindromul (în restul de 50% din cazuri copilul este sănătos). Dacă ambii părinți au o copie mutantă a genei, la 50% copilul dezvoltă sindromul în heterozigoză , în 25% dezvoltă sindromul în homozigoză și, prin urmare, fătul suferă avort spontan , [4] în restul de 25% nu prezintă boală. În cazuri rare, există mutații de novo , care sunt atât de izolate încât nu este posibil să se stabilească apariția lor efectivă în populația generală.
Etiologie

Boala are o moștenire autosomală dominantă , deși microdelecția de novo apare uneori în brațul scurt al cromozomului 7 (care conține locusul genei 7p21, a cărui modificare este responsabilă de sindrom): copiii cu mutație de novo au de obicei mai severe retard mental și anomalii cranio-faciale neobișnuite pentru persoanele cu sindrom. O vârstă înaintată a părinților poate crește riscul acestei mutații cromozomiale. [2]
Analiza genelor asociate și a rearanjării cromozomiale arată că cauza sindromului se găsește într-o mutație a factorului de transcripție TWIST situat pe brațul scurt al cromozomului 7. Această genă codifică o proteină cu o structură elico-buclă. helix care controlează dezvoltarea mezenchimului în timpul dezvoltării, la nivel embrionar, a tubului cranian . Mutațiile pot fi deleții , un nonsens, și mutații / deleții care fie pot scurta sau pauza, sistemul helix-loop-helix necesară pentru a păstra funcționalitatea corectă a proteinelor. Majoritatea indivizilor Saethre-Chotzen au o singură deleție mare la locusul genei 7p21. [5]
S-a observat la șoareci că gena TWIST participă la formarea corectă a craniului, a feței, a membrelor superioare și a membrelor inferioare; șoarecii cu mutații din ambele copii ale genei au fost spontan spălați , în timp ce șoarecii cu mutații dintr-o singură copie a genei au supraviețuit. Investigațiile ulterioare au arătat că porcii de Guineea supraviețuitori aveau defecte moderate ale craniului și picioarelor, foarte asemănătoare cu cele observate în sindromul Saethre-Chotzen la nivelul feței, craniului și membrelor oamenilor. Gena TWIST la șoareci este localizată pe cromozomul 12, care corespunde cromozomului 7 din genomul uman. După aceste descoperiri, cercetătorii au izolat gena TWIST și în cromozomii umani și au elaborat ulterior o hartă genetică care a arătat că ștergerea observabilă la indivizii cu sindromul Saethre-Chotzen este localizată exact pe locusul genei care găzduiește gena TWIST. Până în prezent au fost identificate cinci mutații genetice diferite în cromozomul uman: aceste mutații sunt prezente doar la indivizii cu trăsături fenotipice ale sindromului. Mai mult, studiile efectuate pe insecte din genul Drosophila au arătat că gena TWIST, dacă nu prezintă mutații, efectuează o reglare genică printr-un sistem de interacțiune cu dubla helică a ADN - ului : majoritatea mutațiilor identificate în TWIST modifică sistemele proteice ale atac între genă și ADN, prevenind activarea și reglarea altor gene care ar contribui la dezvoltarea corectă a craniului în perioada fetală. [1]
Clinica
semne si simptome
Gama de semne clinice a sindromului este vastă, iar majoritatea persoanelor cu sindrom prezintă anomalii moderate, cum ar fi defecte faciale asimetrice (unilaterale) și o față aplatizată datorită dezvoltării slabe a orificiilor oculare , oaselor zigomatice și maxilarului . Întârzierea creșterii nu este neobișnuită la cei cu sindrom, ducând adesea la o statură mai mică decât media la vârsta adultă. Dacă multe suturi la nivelul craniului se închid prematur, deformările feței sunt mai severe. [1]
La nivel cranian, brahicefalia și acrocefalia se găsesc de obicei; dolichocefalia este prezentă și în unele cazuri. [6] Distanța orizontală dintre frunte și ceafă este mai mică decât în mod normal. Linia părului tinde să fie foarte înaltă, dând o frunte înaltă și largă. [2]
Atât în mâini și în picioare, este ușor de a găsi fuziunea dintre ele de - a doua și a treia degete ( index - mijloc și illice - trillice, respectiv ). [1] Degetele (chiar și cele care nu sunt afectate de fuziune) tind să fie scurte . Degetul mare și mare deget de la picior sunt adesea extinse și hallux valgus este comună. Există, de asemenea, modificări în pliurile palmei . [2]
La nivel ocular se întâlnesc adesea strabism (datorat anomaliilor mușchilor extraoculari ), hipertelorism , stenoză a canalului lacrimal , ptoză a pleoapei, miopie , epicant , blefarofimoză , atrofie a nervului optic și astigmatism .
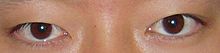

Există, de asemenea, modificări frecvente la nivelul urechilor, nasului și gurii: implantarea urechilor este adesea scăzută, cu rotație înapoi și proeminență a auriculei ; [6] deviația septului nazal duce la un nas „cioc”, ascuțit și ușor îndoit la vârf; pot exista malocluzii datorate cheilor fisurilor , hiperodonția (dinții supranumerari) sau hipoplazia smalțului dentar .
Alte semne ale sindromului, mai puțin frecvente decât cele menționate până acum, sunt statura scurtă, fuziunea vertebrelor , [7] boli cardiace congenitale , dificultăți de limbaj, [8] atrezie anală (datorită unei malformații a rectului ), criptorhidism , [ 6] tulburări de personalitate și diverse anomalii renale . [6]
Cauze și efecte ale craniosinostozei

Neurocraniul este format din trei părți de bază: baza ( osul occipital ), porțiunea anterioară ( osul frontal ) și oasele laterale ( parietale și temporale ) ale capului. Majoritatea oaselor craniene intră deja în contact cu oasele vecine înainte de naștere , cu toate acestea oasele temporale și parietale sunt în mod normal separate una de cealaltă prin suturi , care rămân deschise pentru a asigura o maleabilitate moderată a capului, care facilitează nașterea . Când creierul încetinește dezvoltarea, suturile încep să se închidă, finalizând închiderea în termen de 2 ani de viață.
La pacienții cu sindrom Saethre-Chotzen, însă, sutura coronară care separă oasele temporale de oasele parietale se închide prematur, uneori chiar înainte de naștere. Dacă această închidere are loc doar de o parte sau, în orice caz, într-un mod asimetric, fruntea și fața vor fi la rândul lor asimetrice. Persoanele cu sindrom au capul „ascuțit” ( acrocefalie ) deoarece creierul lor crește mai repede decât craniile; acest lucru duce la o creștere a presiunii intracraniene și, prin urmare, oasele craniene tind să se deplaseze în exterior și în sus pentru a permite expansiunea cerebrală fiziologică. Cele mai mari asimetrii faciale rezultate în urma acestui proces sunt la nivelul ochilor și al obrajilor.
Datorită frunții anormale (înalte și aplatizate), diferitele componente ale feței au un spațiu limitat de dezvoltat; aceasta ia forma unor orificii superficiale ale ochiului (care la rândul lor provoacă hipertelorism ocular) și a pomeților slab pronunțați. Ptoza pleoapelor, frecventă la persoanele cu sindrom, accentuează asimetria generală a feței.
Încercări de laborator și instrumentale
Diagnosticul clinic al sindromului se bazează pe examinarea obiectivă a defectelor structurale la nivel osos; Razele X , tomografia computerizată și rezonanța magnetică nucleară sunt de asemenea utilizate pentru a evalua mai bine deformările. [5]
În genetică moleculară, sindromul poate fi diagnosticat prin analiza genei TWIST care se poate face prin analiza secvenței genomice (care poate identifica deleții și mutații) și prin hibridizare fluorescentă in situ . Analiza secvenței exonului 1 al genei TWIST1 poate oferi indicații cu privire la frecvența mutațiilor: aceasta din urmă poate fi greșită (o singură nucleotidă se schimbă și, prin urmare, acea porțiune a genei codifică un aminoacid diferit), un nonsens (nucleotida mutația are originea unui „codon stop” care blochează transcrierea proteinei), mutații în locul de îmbinare și deleții sau inserții intragenice. Sistemele de analiză de ștergere și duplicare implică reacția în lanț a polimerazei , MLPA (varianta PCR care folosește o singură pereche de primeri în faza inițială) și microarray . Prin hibridizarea fluorescentă in situ este posibilă identificarea translocațiilor și inversiunilor la nivelul regiunii 7p21 a cromozomului 7, care în cazuri rare apare ca un cromozom inelar (acest eveniment cauzează adesea întârzieri mintale la cei afectați de sindrom). [1] [7] Testele citogenetice și testele genetice directe pot fi efectuate pentru a studia în continuare modificările genetice / cromozomiale ale cazului. Testarea genetică directă, în special, poate necesita sânge , păr , piele sau lichid amniotic sau alte țesuturi ale individului cu sindrom suspectat (în special, o extragere de sânge pentru detectarea și studiul ulterior al genei TWIST1 este adesea efectuată în caz de suspiciune diagnostic). [7]
Diagnostic diferentiat
Boala apare într-un diagnostic diferențial cu alte patologii congenitale [1], cum ar fi:
- Sindromul Robinow-Sorauf , caracterizat prin hipertelorism ocular, sept nazal deviat, sindactilie , strabism convergent și epicant .
- Sindromul Muenke , cu hipertelorism ocular, pierderea auzului până la surditate , craniul mărit, pomeții plati și implantarea urechii scăzute.
- Sindromul Crouzon , cu hipertelorism ocular, cap mic și mărit, pierderea auzului, globi oculari proeminenți, nas ascuțit, implantarea urechii joase, strabism, bărbie proeminentă, femur scurt și oase humerus .
- Sindromul Pfeiffer , cu hipertelorism ocular, globi oculari proeminenți, surditate , nas ascuțit, micrognatie și retrognatie .
- Sindromul Apert cu hipertelorism ocular, fruntea proeminentă, ceafa aplatizată, exoftalmie , implantarea urechii joase, fața plană sau concavă, sindactilia și degetele scurte.
- Sinostoză coronariană izolată unilateral , care poate duce la asimetrii faciale similare cu cele ale sindromului Saethre-Chotzen dacă nu este tratată în primele luni de viață.
- Sindrom Baller-Gerold cu microcefalie , exoftalmie , frunte plană, poikilodermie (modificări ale pigmentării pielii), deformare a razei cu un număr redus de degete, întârziere globală a dezvoltării , dezvoltare redusă sau absentă a degetului mare.
Diagnostic precoce
Diagnosticul prenatal al bolii este posibil, dar se face foarte rar; în plus, este fezabilă numai dacă mutația genetică care provoacă boala a fost deja izolată în genomul membrilor familiei care sunt, de asemenea, afectați de sindrom. Testul prenatal se efectuează de obicei în săptămâna 15-18 de sarcină cu colectarea ADN-ului fătului prin amniocenteză , dar poate fi efectuat chiar mai devreme (la 10-12 săptămâni de sarcină) cu CVS . Utilizarea ultrasunetelor este din ce în ce mai frecventă, ceea ce poate detecta anomalii craniene tipice bolii la făt . [6]
Tratament


Anomaliile scheletice ale sindromului sunt de obicei modeste și necesită intervenții chirurgicale minore sau nu necesită deloc intervenție; brahidactilia și sindactilia nu limitează în general funcția degetelor; Sindactilia poate încetini creșterea mâinilor în timpul copilăriei , cu toate acestea această întârziere afectează rareori capacitatea de a utiliza corect mâinile și degetele. Adesea persoanele cu sindrom au degetele de la picioare mărite, deoarece oasele vârfurilor acestor degete tind să se dubleze; acest lucru este observat cel mai frecvent la nivelul degetului mare, dar nu necesită o intervenție chirurgicală corectivă, deoarece nu afectează funcția normală a piciorului și mersul normal.
În cele mai severe cazuri de boală, pe de altă parte, sunt necesare intervenții chirurgicale frecvente și monitorizare constantă în timpul vârstei de dezvoltare: un copil născut cu craniosinostoză coronariană unilaterală cu asimetrie marcată poate fi supus unei operații de cranioplastie în primii ani de viață, pentru a preveni creșterea excesivă a presiunii intracraniene și accentuarea progresivă a asimetriei cranio-faciale. Prin cranioplastie, oasele neurocraniului fuzionate împreună sunt împărțite și apoi fixate din nou într-un aranjament care trebuie să-l urmeze cât mai aproape posibil pe cel tipic la indivizii sănătoși; cranioplastia, prin urmare, are o utilitate preventivă (pentru a reduce creșterea presiunii intracraniene) și o utilitate estetică (pentru a preveni agravarea dismorfiei). În urma operațiilor de cranioplastie, copiii trebuie să poarte o cască cu funcție de protecție și funcție de izolare pentru oasele nou-operate pentru a facilita remodelarea și dezvoltarea normală a acestora; această cască trebuie păstrată în medie aproximativ trei luni. [9]
Structura anormală a craniului poate duce, de asemenea, la erori de refracție mai mult sau mai puțin grave (uneori coroborate cu deteriorarea nervului optic ); cu operații chirurgicale adecvate, orice stenoză a canalului lacrimal poate fi corectată, precum și ptoza pleoapelor (care, dacă nu este tratată, cauzează frecvent ambliopie ). [1]
Chirurgia mezofacială poate fi efectuată în copilărie pentru a reduce orice probleme de înghițire , respirație și malocluzie dentară. Dacă fisura despicată este severă, poate fi corectată printr-o operație care implică și utilizarea unui drenaj transtimpanic . Pentru corectarea defectelor dentare, se utilizează aparate ortodontice care, în cazul persoanelor care suferă de sindromul Shaetre-Chotzen, trebuie implantate după dezvoltarea fizică. [1]
Notă
- ^ a b c d e f g h i Stacey L Blanchford, The Gale Encyclopedia of Genetic Disorders , Michigan, Gale Group, 2002, pp. 1019-1021, ISBN 978-0-7876-5614-0 .
- ^ a b c d Cassidy, Suzanne Allanson, Judith, Management of Genetic Syndromes , New Jersey, John Wiley & Sons, Inc., 2010, pp. 230 -235, ISBN 978-0-470-19141-5 .
- ^ Sindromul Saethre-Chotzen (engleză) , la childrenshospital.org . Adus la 26 ianuarie 2021 (arhivat din original la 16 septembrie 2013) .
- ^ Sindromul Saethre-Chotzen , la seattlechildrens.org . Adus la 26 ianuarie 2021 .
- ^ a b Galie M. Clauser L., Sindromul Saethre-Chotzen (engleză) ( PDF ), la orpha.net , Orphanet. Adus la 28 octombrie 2012 .
- ^ a b c d e James Wynbrandt, Tulburări genetice și defecte congenitale , New York, Facts on File, Inc., 2008, pp. 340 , ISBN 978-0-8160-6396-3 .
- ^ a b c Gallagher E, Ratisoontorn C, Cunningham M, https://www.ncbi.nlm.nih.gov/books/NBK1189/ , în Sindromul Saethre Chotzen (engleză) , NCBI, 1993.
- ^ Sindromul Saethre-Chotzen (engleză) , la childrensomaha.org , Spitalul pentru copii și Centrul medical. Accesat la 25 octombrie 2012 .
- ^ Opțiuni chirurgicale pentru craniosinostoză (engleză) , la hopkinsmedicine.org , Johns Hopkins Medicine. Adus la 28 noiembrie 2012 .