Distrofia miotonică
| Distrofia miotonică | |
|---|---|
| Boala rara | |
| Cod. SSN | RFG090 |
| Specialitate | neurologie |
| Etiologie | mutaţie |
| Clasificare și resurse externe (EN) | |
| OMIM | 160900 |
| Plasă | D009223 |
| GeneReviews | Prezentare generală și Prezentare generală |
| Sinonime | |
| Miotonia distrofică | |
| Eponime | |
| Hans Gustav Wilhelm Steinert | |

Distrofia miotonică (DM) este o boală neuromusculară genetică degenerativă cu caracter autosomal dominant, caracterizată printr-un tablou clinic foarte variabil și un curs lent progresiv, al cărui debut poate apărea la orice vârstă. Reprezintă a doua formă cea mai frecventă de distrofie musculară după distrofia musculară Duchenne .
Tabloul clinic se caracterizează prin pierderea masei musculare , mioton , cataractă , defecte ale sistemului de conducere cardiacă , alterări endocrine și deficite cognitive în cazurile congenitale infantile de DM1. Există un fenomen de anticipare prin care debutul tinde să aibă loc la o vârstă tot mai tânără, din generație în generație, în aceeași familie.
Pacienții cu distrofie miotonică de tip 1 sau Steinert, cu excepția formei tardive, au o speranță de viață mai scurtă, fiind predispuși la aritmie severă și cardiomiopatie (care poate duce la moarte subită cardiacă ) și la tulburări respiratorii grave. În distrofia miotonică de tip 2 sau Ricker, speranța de viață a pacientului poate fi, de asemenea, normală.
Epidemiologie
Prevalența a fost calculată la aproximativ 1 din 8.000 de persoane. [1] [2]
Clasificare
Există diferite forme de distrofie miotonică recunoscute prin examinarea ADN:
- Distrofia miotonică de tip 1 (DM1), numită și boala Steinert, identificată în 1909 [3], care poate avea un debut precoce la sugari și copii.
- Distrofia miotonică de tip 2 (DM2) sau PROMM (miopatia miotonică proximală), numită și boala Ricker
Aproximativ 98% din toate cazurile de distrofie miotonică se încadrează în tipul 1, cu toate acestea tipul 2 se caracterizează prin prezentare atipică și fenotipuri neobișnuite, cu simptome diferite de forma clasică, prin urmare este probabil ca diagnosticul să fie subestimat.
Specialiștii investighează în prezent existența altor tipuri de distrofie miotonică.
Genetica
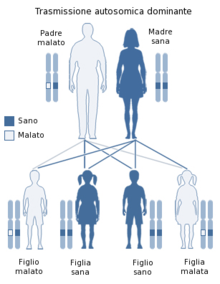
Distrofia miotonică este o tulburare genetică autosomală dominantă, prin urmare se dezvoltă în urma dobândirii genei afectate de la un părinte. Probabilitatea de a moșteni gena afectată este de 50%.
DM se caracterizează prin prezența tripletelor de nucleotide repetate. Repetițiile perechilor sau tripletelor de nucleotide sunt frecvente în ADN, dar în această patologie repetările sunt în număr exagerat în comparație cu repetările prezente în ADN-ul normal; acest fenomen se numește „ amplificare ”. În ultimii ani, au fost identificate numeroase boli cu transmitere genetică în care mecanismul de deteriorare este legat de prezența repetării tripletelor, prima care a fost studiată și cea mai cunoscută a fost coreea Huntington, care împărtășește caracteristica debutului cu distrofia miotonică mai devreme cu fiecare succesiv. generaţie.
DM1
În forma DM1 (boala Steinert) gena mutantă se numește DMPK ( proteina kinază distrofică miotonică ) și codifică o miozin kinază exprimată în mușchiul scheletic. Această genă este localizată în brațul lung al cromozomului 19 (în poziția 19q13.3). In acest locus exista un defect molecular specific, constând dintr - o secvență trinucleotidă instabilă ( Citosin timină Guanine ) în 3'UTR care se repetă de 50-4000 ori când în populația normală intervalul variază între 5 și 37 de ori.
DM2
În forma DM2 (numită și miopatie miotonică proximală / PROMM sau boala Ricker) există în mod similar un defect în gena ZNF9 pe cromozomul 3 la poziția 3q21. Numărul de repetări variază de la 75 la peste 11.000, dar în acest caz nu pare să existe nicio diferență în gravitatea bolii sau în precoce de debut. Fenomenul anticipării în această formă pare a fi mai puțin semnificativ și doar o ușoară anticipare este raportată în literatura de specialitate. În acest caz, repetarea implică patru nucleotide. [4]
Tablou clinic
Tabloul clinic este variabil, frecvent în DM1 constatarea miopatiei , disartriei , atrofiei , hipotiroidismului , întârzierii mintale (numai dacă debutul infantil), implicarea miocardului [5] poate fi foarte gravă, ducând la forme de cardiomiopatie difuză . [6] Formele congenitale și infantile sunt severe, în timp ce formele juvenil-preadolescent și adult sunt mai puțin. Forma mai puțin severă este cea târzie.
Nistagmus , [7] disfagie și dureri abdominale [8] sunt frecvent observate în DM2, în timp ce tulburări ale spectrului autist, inclusiv sindromul Tourette, sunt observate în DM1. [9]
Pacienții cu DM2 prezintă dureri musculare, oboseală, rigiditate, slăbiciune în mușchii proximali ai membrului inferior (coapsa). [10] Forma mai puțin severă se numește miopatie miotonică proximală.
DM prezintă deseori probleme endocrine ( hipotiroidism și hipertiroidism , diabet zaharat ), cataractă precoce, probleme gastro-intestinale, hipersomnie , tulburări psihiatrice, probleme respiratorii.
Distribuția slăbiciunii musculare este diferită pentru cele două forme: în DM1 sunt afectate masele musculare ale feței și maxilarului , cu ptoză pleoapelor , slăbiciune a mușchilor gâtului , mâinilor și a părții distale a piciorului ( piciorului ). În DM2 slăbiciunea este mai evidentă în mușchii proximali, deci în apropierea trunchiului: ceafă, umeri, flexori ai șoldului și picioarele superioare.
Diagnostic
Diagnosticul distrofiei miotonice poate fi dificil, deoarece implică diagnosticul diferențial cu boli neuromusculare care împărtășesc unele aspecte ale tabloului clinic. Patologiile neuromusculare sunt în mare parte rare și în prezent sunt cunoscute peste 40 care devin 100 dacă se iau în considerare subtipurile. Prin urmare, un pacient cu o imagine clinică complexă care ar putea fi afectată de DM va trebui să fie trimis la un specialist, dar în funcție de simptomul de debut, pacientul ar putea fi direcționat către un neurolog, un cardiolog, un oftalmolog, un endocrinolog sau un reumatolog . Deoarece prezentarea este frecvent atipică, este posibil ca, dacă nu este consultat un expert în boli neuromusculare, diagnosticul nu va fi pus.
Deși în prezent nu există tratamente specifice pentru DM și terapia se bazează pe intervenții simptomatice (problemele se confruntă pe măsură ce apar), este important ca diagnosticul să fie formulat corect, atât pentru a monitoriza pacientul, cât și pentru a putea recunoaște cele mai grave și manifestări potențial fatale (de exemplu, probleme cardiace), atât pentru a oferi consiliere genetică cu privire la riscul ridicat de transmitere la descendenți.
Riscul anestezic este de așa natură încât prezența DM trebuie raportată la fiecare vizită medicală, chiar și pentru probleme care nu au legătură cu patologia.
Diagnosticul certitudinii se face prin examinarea ADN-ului.
Terapie
Nu există tratamente eficiente împotriva acestei forme de distrofie musculară , mexiletina se administrează pentru combaterea parțială a miotoniei (doze de 75 mg - 150 mg). [11]
Accentul este pus pe gestionarea complicațiilor bolii, în special a celor legate de sistemul cardiopulmonar, deoarece acestea reprezintă 70% din decesele cauzate de DM1. [12] Poate fi necesară inserția stimulatorului cardiac pentru persoanele cu anomalii ale conducerii cardiace. Îmbunătățirea calității vieții care poate fi măsurată folosind chestionare specifice [13] este, de asemenea, un obiectiv principal al îngrijirii medicale. Apneea centrală a somnului sau apneea obstructivă a somnului poate provoca somnolență excesivă în timpul zilei, iar aceste persoane ar trebui să fie supuse unui studiu de somn. Ventilația neinvazivă poate fi oferită în caz de anomalii respiratorii.
Unele studii mici au sugerat că imipramina, clomipramina și taurina pot fi utile în tratamentul miotoniei. [12] Cu toate acestea, din cauza dovezilor slabe și a efectelor secundare potențiale, cum ar fi aritmiile cardiace, aceste tratamente sunt rareori utilizate. Un studiu recent din decembrie 2015 a arătat că un antibiotic comun aprobat de FDA, eritromicina , a redus miotonia la șoareci. [14] Studiile la om sunt planificate pentru eritromicină. Eritromicina a fost utilizată cu succes la pacienții cu probleme gastrice. [15]
Splicarea modificată a canalului specific 1 muscular (ClC-1) s-a dovedit a provoca fenotipul miotonic al DM1 și este reversibilă la modelele de șoareci care utilizează antisens morfolin pentru a modifica splicarea ARNm ClC-1. [16]
Prognoză
Prognosticul este slab în DM1 (cu excepția DM1 târziu); riscul de deces este mai mare la tipul 1, deoarece este mai predispus la aritmii fatale care cauzează moartea subită din torsada vârfurilor. DM2 este mai puțin predispus la moarte subită cardiacă decât DM1 și cardiomiopatie .
Forma tardivă a DM1 este uneori asimptomatică, în timp ce forma miopatică miotonică proximală a DM2 are o afectare cardiacă rară și un prognostic mai bun. [17]
Speranța medie de viață a unui pacient este în general mai mică, dar poate fi normală în cele două forme mai ușoare, în special în DM2. [18]
Notă
- ^ Antonio Cao, Boli genetice. Molecule și gene. Diagnostic, prevenire și terapie , Padova, Piccin, februarie 2004 [2004] .
- ^ (EN) Genetics Home Reference,distrofia miotonică , pe Genetics Home Reference. Adus la 10 decembrie 2018 .
- ^ Steinberg H, Wagner A., Hans Steinert: 100 de ani de distrofie miotonică , în Cardiol Prat. , vol. 79, august 2008, pp. 961-70.
- ^ Liquors CL, Ricker K, Moseley ML și colab. , Distrofia miotonică de tip 2 cauzată de o expansiune CCTG în intronul 1 al ZNF9 , în Science (jurnal) , vol. 293, nr. 5531, august 2001, pp. 864–7, DOI : 10.1126 / science.1062125 , PMID 11486088 .
- ^ Sá MI, Cabral S, Costa PD, Coelho T, Freitas M, Torres S, Gomes JL., Cardiac implicent în distrofia miotonică de tip 1 , în Rev Port Cardiol. , vol. 27, 2007, pp. 829-840.
- ^ McDonnell M, Alcantar J, Wachsner RY, Meymandi SK., Cardiomiopatie și emboli pulmonari multipli la un pacient cu distrofie miotonică , în Insuficiența cardiacă congestivă. , vol. 14, 2008, pp. 106-110.
- ^ Ajroud-Driss S, Sufit R, Siddique T, Hain TC., Implicarea oculomotorie în distrofia miotonică de tip 2. , în nervul muscular. , 2008.
- ^ Tieleman AA, van Vliet J, Jansen JB, van der Kooi AJ, Borm GF, van Engelen BG., Implicarea gastro-intestinală este frecventă în distrofia miotonică de tip 2. , în tulburarea neuromusculară. , vol. 18, august 2008, pp. 646-649.
- ^ Ekström AB, Hakenäs-Plate L, Samuelsson L, Tulinius M, Wentz E .., Condiții ale spectrului autismului în distrofia miotonică de tip 1: un studiu pe 57 de indivizi cu forme congenitale și de copilărie. , în Am J Med Genet B Neuropsihiatr Genet. , 147B, 2008, pp. 918-926.
- ^ (Day & al, 2003).
- ^ Research Laboratories Merck, Manualul Merck ediția a cincea Pagina 2600 , Milano, Springer-Verlag, 2008, ISBN 978-88-470-0707-9 .
- ^ a b ( EN ) Chris Turner și David Hilton-Jones, Distrofiile miotonice: diagnostic și management , în Journal of Neurology, Neurosurgery & Psychiatry , vol. 81, nr. 4, 1 aprilie 2010, pp. 358–367, DOI : 10.1136 / jnnp.2008.158261 . Adus la 11 iunie 2020 .
- ^ (EN) Antoine Dany și Amandine Coralie Barbe Rapin, Construirea unui chestionar privind calitatea vieții pentru boala neuromusculară lent progresivă , în Quality of Life Research, vol. 24, n. 11, 1 noiembrie 2015, pp. 2615-2623, DOI : 10.1007 / s11136-015-1013-8 . Adus la 11 iunie 2020 .
- ^ Masayuki Nakamori, Katarzyna Taylor și Hideki Mochizuki, Administrarea orală de eritromicină scade toxicitatea ARN în distrofia miotonică , în Annals of Clinical and Translational Neurology , vol. 3, nr. 1, 10 decembrie 2015, pp. 42–54, DOI : 10.1002 / acn3.271 . Adus la 11 iunie 2020 .
- ^ (EN) Rönnblom A., S. Andersson și Hellström PM, Golirea gastrică în distrofia miotonică , în European Journal of Clinical Investigation, vol. 32, nr. 8, 2002, pp. 570–574, DOI : 10.1046 / j.1365-2362.2002.01028.x . Adus la 11 iunie 2020 .
- ^ Thurman M. Wheeler, John D. Lueck și Maurice S. Swanson, Correction of ClC-1 splicing elimină canalopatia clorurii și miotonia în modele de șoarece de distrofie miotonică , în The Journal of Clinical Investigation , vol. 117, nr. 12, 3 decembrie 2007, pp. 3952–3957, DOI : 10.1172 / JCI33355 . Adus la 11 iunie 2020 .
- ^ Miopatie miotonică proximală
- ^ Groh WJ, Groh MR, Saha C, Kincaid JC, Simmons Z, Ciafaloni E, Pourmand R, Otten RF, Bhakta D, Nair GV, Marashdeh MM, Zipes DP, Pascuzzi RM., Anomalii electrocardiografice și moarte subită în tipul de distrofie miotonică 1. , în N Engl J Med. , Vol. 358, iunie 2008, pp. 2688-2697.
Bibliografie
- Joseph C. Segen,Dicționar concis de medicină modernă , New York, McGraw-Hill, 2006, ISBN 978-88-386-3917-3 .
- Douglas M. Anderson, A. Elliot Michelle, Mosby's medical, nursing, and Allied Health Dictionary ediția a șasea , New York, Piccin, 2004, ISBN 88-299-1716-8 .
Elemente conexe
Alte proiecte
-
 Wikimedia Commons conține imagini sau alte fișiere despre distrofia miotonică
Wikimedia Commons conține imagini sau alte fișiere despre distrofia miotonică
linkuri externe
- Baza de date căutabilă la Neuromuscular Research olandeză
- Al 140-lea atelier internațional ENMC 2006 Distrofia miotonică DM2 / PROMM și alte distrofii miotone]
- Informații despre boli de la Fundația de distrofie miotonică
- Informații de la Organizația Internațională a Distrofiei Miotone
- Informații MDSG , pe mdsguk.org . Adus la 26 august 2009 (arhivat din original la 29 decembrie 2008) .
- Informații de la Centrul pentru boli neuromusculare
- DM Toolbox Instrumente de cercetare pentru distrofia miotonică de la Fundația Marigold

| Controlul autorității | Thesaurus BNCF 43723 · LCCN (RO) sh85089286 · BNF (FR) cb12268952p (data) · BNE (ES) XX4435101 (data) |
|---|