Sindromul Prader-Willi
| Sindromul Prader-Willi | |
|---|---|
 | |
| Boala rara | |
| Cod. SSN | RN1310 |
| Specialitate | genetică clinică , pediatrie și neurologie |
| Clasificare și resurse externe (EN) | |
| OMIM | 176270 |
| Plasă | D011218 |
| MedlinePlus | 001605 |
| eMedicină | 947954 |
| GeneReviews | Prezentare generală |
| Eponime | |
| Andrea Prader Heinrich Willi | |
Sindromul Prader Willi (prescurtat PWS: sindromul Prader Willi ) este o boală genetică rară (afectează 1 din 15.000-25.000 de nașteri vii), caracterizată prin modificarea cromozomului 15. Își ia numele de la primul care l-a identificat în 1956 : Andrea Prader , Heinrich Willi , Alexis Labhart, Andrew Ziegler și Guido Fanconi la Clinica de pediatrie a Universității Zurich din Elveția . Tulburările apar din copilărie și se alternează treptat, în principal din cauza tulburărilor hormonale care cauzează o obezitate îngrijorătoare în adolescență.
Etiologie
Prader-Willi este cel mai frecvent dintre sindroamele de micro- deleție cromozomiale . Apare pentru două cauze diferite stabilite, ambele de tip genetic:
- Ștergerea unei regiuni, totală sau parțială, pe cromozomul 15 de origine paternă. Această regiune specială este supusă amprentării părintești și este activă în cromozomul patern, în timp ce este inactivă în cea maternă.
- Disomia maternă uniparentală: prezența a două copii de origine maternă pe ambii cromozomi 15, ambele rezultând inactive, chiar dacă pot fi identice sau diferite.
Ștergerea de la imprimare
Ce este imprimarea
Imprimarea este un fenomen fiziologic al celulelor somatice . Într-o celulă somatică există 46 de cromozomi „egali” la doi, deci două copii ale fiecărei gene , fiecare dintre acestea provin de la un singur părinte. Pentru fiecare genă avem o copie de la tată și una de la mamă. Acestea sunt ușor diferite unele de altele, ca fenomen de polimorfism , dar ambele codifică pentru aceeași proteină. Deoarece este suficientă o singură copie a majorității genelor, acest proces are loc: imprimarea tace una dintre cele două gene (este compactată cu metilare pentru a nu da acces la proteinele transcripționale), iar ceea ce este transcris activ este doar unul., Care aleatoriu vine fie de la mamă, fie de la tată. Acest fenomen apare în cursul gametogenezei. Majoritatea genelor, imediat după fertilizare (stadiul postzigotic), suferă un val de demetilare care afectează aproape întregul genom. Genele imprimate sunt excluse din acest fenomen, tocmai pentru că metilarea în acest caz servește la indicarea originii parentale a genei.
Cazul patologic al PWS
În PWS, gena maternă este redusă la tăcere deoarece este imprimată, în timp ce cea paternă este ștearsă. Gena în cauză provine de la cromozomul 15, în regiunea 15q11-q13. În această patologie, contribuția paternă lipsește, prin urmare, și vor exista o serie de tulburări care decurg din lipsa proteinelor care derivă din aceasta. PWS este strâns legat de sindromul Angelman ( AS ), care este cauzat de amprentarea paternă și de ștergerea genei materne. Simptomele sunt foarte diferite și există studii în acest sens, pentru a defini modul în care originea este identificată și poate varia radical.

Locusul 15q11-q13
Acest locus conține gene care sunt supuse amprentării specifice materne sau paterne și dintre care doar una este exprimată. Regiunea ștearsă conține informațiile care codifică proteina umană necdin sau necdin (locus NDN), aproape de regiunea centromerică a deleției, între cele două gene ZNF127 și SNRPN, ambele imprimate. Necdinul este o proteină nucleară care este exprimată doar de anumiți neuroni din creier ( SNC ). Se pare (din studii încă în desfășurare pe șoareci) că guvernează arestarea permanentă a creșterii celulare după perioada embrionară mitotică, în perioada de așezare neuronală. Deși cercetările anterioare de cartografiere genetică pe necdină l-au identificat pe cromozomul 7, s-a demonstrat că expresia este limitată la prezența alelei paterne în ARN-ul creierului neonatal. Expresia nu se găsește numai în creier, ci și în alte țesuturi, dar cu cele mai înalte niveluri în placentă și sistemul nervos central. NDN este exprimat exclusiv de alela paternă la fibroblastele umane. Aceste observații conduc la clarificarea modului în care pierderea alelei paterne poate da naștere la tulburări neurologice ale persoanelor cu PWS.
Actualizări de cercetare
Cercetări recente indică înțelegerea modului în care metilarea distinge alelele materne și paterne. Genele SNRPN, MKRN3 și NDN au fost identificate și studiate și s-a constatat că sunt exprimate numai de alela moștenită de la tată. Prin urmare, pacienților cu PWS le lipsește cele de mai sus.
- SNRP este implicat în îmbinarea pre- ARNm (transcript primar);
- MKRN3 codifică o proteină deget de zinc ;
- NDN (vezi mai sus)
Toate aceste gene au o insulă CpG '5', care nu este metilată în alela paternă exprimată, în timp ce este metilată în alela imprimată (maternă). O constatare importantă este că semnalul de imprimare pentru gena SNRP începe deja în gametogeneza masculină și feminină. Informații suplimentare despre acest lucru: [1]
Disomie uniparentală
A doua cauză se numește disomie uniparentală, adică atunci când perechea de cromozomi 15 nu mai este compusă dintr-un membru cu material matern și unul cu material patern, ci de 2 membri materni sau paterni, pierzându-se în consecință tot patrimoniul genetic de tip patern sau matern. . În cazul acestui sindrom, cromozomul matern 15 este prezent în copie dublă, în timp ce dacă ați avea o copie dublă a cromozomului paternal 15, atunci ați avea sindromul Angelman .
Analiza de laborator
Analiza de laborator se efectuează în caz de:
- Cuplurile care au un copil cu PWS sau AS și așteaptă altul
- Suspectiv în timpul gestației (vezi în Simptome)
- Simptome vizibile ale sugarului sau copilului
Dintr-o probă de sânge , limfocitele sunt cultivate și analizate prin liza celulară în soluție hipotonică și izolarea ADN-ului .
Testele de biologie moleculară s-au dovedit a fi foarte utile pentru un diagnostic precoce corect și pentru a stabili cea mai potrivită terapie.
Test de metilare
Se efectuează o Southern Blot care constă în hibridizarea ADN-ului pacientului (după tratamentul cu enzime de restricție ) cu ADN „sondă” marcat în zonele critice care trebuie evidențiate. Dacă hibridizarea sau împerecherea dintre ADN-ul sondei și cel al pacientului este aproape perfectă, pacientul nu este afectat; dacă, pe de altă parte, prezintă malmatch-uri, înseamnă că există mutații genetice la pacient. Această tehnică nu distinge diferitele mutații, dar este tehnica preferată, deoarece este mai ieftină decât altele și vă permite să diagnosticați / excludeți PWS la 100%.
MSPCR
PCR specifică pentru metilare este o tehnică mai specifică pentru recunoașterea PWS. Imprimarea este o compactare a unor zone specifice datorită metilării citozinelor . Cu acest test, ADN-ul este tratat cu bisulfit de sodiu , care schimbă numai citozinele nemetilate în uracil . Un ciclu PCR (duplicarea selectivă a fragmentelor de ADN), cu primeri care se disting în metilat și nemetilat.
PEŞTE
Hibridizarea fluorescentă in situ (Fluorescent In Situ Hybridization sau FISH): permite examinarea exactă a sitului compromis prin utilizarea sondelor specifice pentru regiunea conjugată cu molecule fluorescente. Hibridizarea acestor sonde (individ normal) sau non-hibridizare (individ cu patologie) este detectată prin microscop optic cu fluorescență.
Simptomatologie
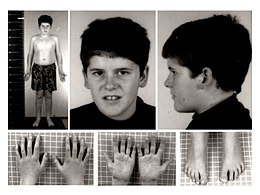
Cei născuți cu acest sindrom prezintă imediat o hipotonie marcată care dispare treptat odată cu adolescența. Ulterior, de la doi la șase ani, acești copii își pot dezvolta un apetit de nesatiat din cauza unei disfuncții a hipotalamusului care îi va însoți pe tot parcursul vieții. Unele dintre ele prezintă o întârziere mintală care poate fi ușoară sau severă în funcție de individ, hipogonadism , strabism, mâini și picioare mici. Cu toate acestea, hiperfagia este cea mai gravă problemă; de fapt, dacă nu este controlat cu un regim alimentar strict, poate duce la obezitate severă, cu toate problemele care decurg din aceasta (vasculară, diabet etc.) până la compromiterea sănătății subiectului însuși. Copiii PW sunt ajutați cu administrarea hormonului de creștere GH , care asigură o mai mare vitalitate, îi face să crească corect și limitează disfuncțiile metabolice care îi împing să se îngrașe mai ușor decât subiecții sănătoși.
Simptome generale
Simptomele descrise de Prader și aa. în 1956:
- scăderea activității fetale
- hipotonie infantilă severă
- probleme alimentare cu copilăria
- hipogonadism hipogenitalism
- întârzierea vârstei osoase și statura scurtă
- mâini și picioare mici
- întârziere mentală moderată
- facies caracteristic
- obezitate (copilărie timpurie)
- probleme de comportament (adolescență)
- tendință de a dezvolta diabet (adolescență)
Simptome majore
- Central neonatală și infantil hipotonie pisto slab , care imbunatateste cu varsta.
- Probleme de alimentație în copilăria timpurie, creștere slabă în greutate.
- Obezitate centrală după an, dar înainte de vârsta de 6 ani.
- Caracteristici somatice caracteristice: dolichocefalie în copilărie, diametru bifrontal îngust, ochi în formă de migdale, gură mică cu buza superioară subțire, colțurile gurii orientate în jos (cel puțin 3 semne), marginea pleoapei orientată în jos.
- Hipogonadism
- Dezvoltarea psihomotorie întârziată înainte de vârsta de 6 ani;
- întârziere mintală ușoară până la moderată sau dizabilități de învățare la copiii mai mari. (retard mental: 63% ușor, 31% mediu, 6% sever)
- Hiperfagia / furtul alimentelor / obsesia alimentelor.
- Anomalii citogenetice sau moleculare la analiza regiunii 15q11-q13.
Simptome minore
- Reducerea mișcărilor fetale, plâns slab la sugar rezolvându-se progresiv
- Caracteristici comportamentale: excese de furie, izbucniri violente și comportament obsesiv-compulsiv
- Tulburări de somn și apnee în somn
- Mic de statura
- Hipopigmentare
- Mâini mici (<25 cent.) Și / sau picioare mici (<cent. 10).
- Mâinile încleștate cu marginea ulnară dreaptă
- Anomalii oculare (exotropie, miopie)
- Saliva groasă, lipicioasă, cu cruste în colțurile gurii
- defecte în articularea vorbirii
- Culegerea pielii
Alte simptome
- Prag de durere ridicat
- Scăderea reflexului gag
- Modificări ale termoreglării la prima copilărie sau sensibilitate modificată la temperatură la a 2-a copilărie
- Scolioză și / sau cifoză
- Adrenarhia timpurie
- Osteoporoza
- Jocuri de îndemânare în răbdare (puzzle-uri)
- Investigații neuromusculare normale
Tratament
Tratamentul de abilitare, în ceea ce privește vârsta pediatrică, se bazează pe ședințe de fizioterapie (în fața afecțiunii hipotonului frecvent), logopedie (pentru a face față întârzierii limbajului și a dispraxiei buco-fonatorii), ședințe educaționale și psihomotorii (pentru a face față la condiția de întârziere mintală, dobândind astfel premisele învățării pentru intrarea în școala primară și pentru a gestiona vorbirea de hipotonie și dispraxie prezente pentru a încuraja coordonarea motorie a copilului).
Bibliografie
- Giovanni Neri, Maurizio Genuardi, Genetica umană și medicală , Elsevier, 2010, ISBN 88-214-3172-X .
Elemente conexe
Alte proiecte
-
 Wikimedia Commons conține imagini sau alte fișiere despre sindromul Prader-Willi
Wikimedia Commons conține imagini sau alte fișiere despre sindromul Prader-Willi
linkuri externe
- ( EN ) Sindromul Prader-Willi , în Encyclopedia Britannica , Encyclopædia Britannica, Inc.
- Federația Națională a Sindromului Prader Willi , pe praderwilli.it .

| Controlul autorității | Tezaur BNCF 67274 · LCCN (EN) sh85106050 · GND (DE) 4201277-6 · BNF (FR) cb12468434q (dată) · NDL (EN, JA) 01.179.976 |
|---|