Neurooncologie

| Tumori ale sistemului nervos central |
|---|
Familiile de cancer conform clasificării OMS din 2007 .
|
Neuro-oncologia este o ramură a medicinei care se ocupă cu studiul, diagnosticul și tratamentul tumorilor sistemului nervos .
În special, această disciplină este interesată de tratamentul neurologic, medical, chirurgical și oncologic al pacienților care suferă de neoplasme primare sau metastatice ale sistemului nervos central , periferic și orice altă afecțiune sau complicație legată de sistemul nervos de origine neoplazică sau prin tratamente efectuate pentru tratarea acestui tip de boală. [1] [2] [3] Prezenta discuție se referă la principalele familii de tumori ale sistemului nervos central. Vă rugăm să consultați alte elemente pentru analiza tumorilor sistemului nervos periferic. În mod similar, expunerea la complicații este abordată în cadrul intrării referitoare la tumora anume sau la familia tumorii.
Tumori ale sistemului nervos central - Tumori primare
În literatură, tumorile sistemului nervos central (SNC) au o primă subdiviziune dură în primar (adică originar din SNC) și metastatic (originar din alt organ ). Dintre aceștia din urmă, chiar dacă au o incidență egală cu de aproximativ 10 ori prima, [4] vom menționa la final, referind discuția extinsă la alte elemente specifice.
Termenul de tumori cerebrale este foarte des folosit pentru a se referi generic la neoplasmele care se dezvoltă în craniu , chiar dacă în mod necorespunzător. De exemplu, meningioamele , care se comprimă, dar invadează rareori creierul , sunt considerate în continuare tumori cerebrale, la fel ca și tumorile glandelor hipofizare și pineale , care nu sunt strict parte a creierului, dar locuiesc în teca . Denumirea mai adecvată ar fi cea a tumorilor intracraniene . [5] În cele ce urmează vom folosi în mod normal adjectivul „cerebral”; termenul „intracranian” va fi folosit mai rar sau acolo unde există riscul de ambiguitate.
| Tab. 1 - Distribuția tumorilor SNC pe subtip histologic CBTRUS 1998-2002 (N = 63.968) [6] | |
| Histologie | Procent |
| Glioblastom * | 20.3 |
| Astrocitom * | 9.8 |
| Oligodendrogliom * | 3.7 |
| Ependimom * | 2.3 |
| Tumori embrionare (inclusiv meduloblastom) | 1.7 |
| Tumori ale tecii nervului periferic | 8.0 |
| Meningiom | 30.1 |
| Limfom | 3.1 |
| Craniofaringiom | 0,7 |
| Tumori ale hipofizei | 6.3 |
| Alții | 14.0 |
| (*) În SNC, glioamele reprezintă aproximativ 40% din toate tumorile și aproximativ 78% din tumorile maligne. | |
Tumorile primare ale sistemului nervos central (Tabelul 1) [6] cuprind un set divers de entități patologice, fiecare cu istoria sa naturală distinctă. Pentru faptul că tumorile gliei constituie singure aproape 40% din astfel de tumori, poate opera o primă distincție între tumorile gliale ( glioame ) și tumorile non-gliale.
Cele mai frecvente gliome sunt astrocitoamele (provenite din celulele astrocitice ale gliei), oligodendrogliomele (din celulele oligodendrogliale ) și ependimoamele (din celulele ependimale ). Tabelul 2 raportează greutatea fiecărui element din familia gliomilor.
| Tab. 2 - Distribuția glioamelor după subtip histologic CBTRUS 1998-2002 (N = 25.539) [6] | |
| Histologie | Procent |
| Glioblastom * | 50,7 |
| Astrocitom anaplastic * | 7.9 |
| Astrocitom difuz * | 1.7 |
| Astrocitom pilocitar * | 5.7 |
| Alte astrocitoame * | 9.1 |
| Oligodendrogliom | 9.2 |
| Ependimom | 5.6 |
| Alții | 10.1 |
| (*) Astrocitoamele (inclusiv glioblastomul) reprezintă aproximativ 75% din toate glioamele. | |
Morfologia și caracteristicile genetice ale fiecărui tip de gliom vor fi evidențiate în secțiunile dedicate fiecărui tip particular de neoplasm. Este util să se anticipeze o defalcare între gliomuri circumscrisă și Glioamele difuze. Exemple ale primului grup sunt următoarele variante astrocitice destul de neobișnuite: astrocitom pilocitar , xanthastrocitom pleomorf și astrocitom cu celule gigant subependimale . Majoritatea glioamelor au caracteristici de difuzie ridicată în substanța albă , ceea ce face practic imposibilă îndepărtarea lor prin intervenție chirurgicală. Neoplasmele non-gliale au atât histologie benignă (cum ar fi, de obicei, meningioamele și adenoamele hipofizare), cât și maligne, cum ar fi tumorile neuroectodermice primare și meduloblastoamele, limfoamele primare ale SNC și tumorile destul de rare ale celulelor germinale. [4]
Epidemiologie

Tumorile maligne primare ale sistemului nervos central sunt relativ rare, reprezentând aproximativ 2% din toate neoplasmele non-benigne. Cu toate acestea, tumorile cerebrale, atât benigne cât și maligne, sunt o sursă neobișnuită de morbiditate și mortalitate ridicată , datorită și particularităților organelor intracraniene. În Statele Unite, aproximativ 43 800 de cazuri noi de tumori cerebrale sunt diagnosticate anual, dintre care 3410 implică copii și adolescenți. Dintre toți acești pacienți noi, aproximativ 12.760 vor muri. Incidența tumorilor cerebrale este de 14,8 cazuri noi pe an la 100.000 de indivizi, dintre care aproximativ jumătate sunt benigne din punct de vedere histologic. Acestea din urmă, dacă nu pot fi tratate chirurgical sau prin radioterapie , pot fi fatale datorită creșterii progresive în spațiul închis al craniului. Femeile au o incidență ușor mai mare decât bărbații (15,1 față de 14,3 cazuri noi pe an la 100 000 de indivizi), probabil din cauza incidenței mai mari a meningioamelor la femei. Tumorile maligne ale sistemului nervos central sunt principala cauză de deces prin tumori solide la copii și a treia cauză principală de deces prin cancer la adolescenți și adulți tineri cu vârste cuprinse între 15 și 34 de ani. [4] [7]
În tabelul 1, care listează principalele tumori ale SNC, se observă că meningiomul este cea mai frecventă tumoare intracraniană benignă și glioblastomul cel mai frecvent cu histologie malignă. [6] [8]
Etiologie

O predispoziție genetică la malignitatea sistemului nervos central este relativ neobișnuită, deși unele glioame pot apărea ca complicații ale mai multor boli familiale.
În special, mutația unor gene supresoare tumorale caracterizează mai multe sindroame ereditare, care arată o susceptibilitate crescută la dezvoltarea tumorilor cerebrale. Neurofibromatoza de tip 1 (mutația genei NF1 ), sindromul Turcot (mutația APC ), sindromul Gorlin (mutația PTCH ) [9] și sindromul Li-Fraumeni (mutația TP53 sau CHEK2 ) sunt asociate cu un risc mai mare de a dezvolta tumori cerebrale.
Factorii de mediu legați de tumorile cerebrale primare sunt dificil de identificat. În unele studii, expunerea la clorură de vinil a fost asociată cu o incidență crescută a glioamelor de grad înalt (în ceea ce privește conceptul de gradare a se vedea mai jos).
Radiațiile ionizante sunt singura cauză rară, bine identificată, a tumorii cerebrale primare. În special, tratamentul cu radioterapie a copiilor cu tinea capită , adică pacienții culeucemie limfocitară acută , craniofaringiom sau limfom non-Hodgkin a fost asociat cu un risc crescut de gliom. În cele din urmă, există un risc crescut de limfom cerebral primar (dar nu și de alte tipuri de cancer al sistemului nervos central) la pacienții cu SIDA . [10]
Clinica
semne si simptome
Simptomatologia neoplaziei cerebrale este cauzată de efectul de masă, infiltrarea parenchimatoasă și distrugerea țesuturilor.
Cefaleea , cel mai frecvent simptom, este legată de efectul de masă și afectează aproximativ 35% dintre pacienți. Apariția durerilor de cap severe la un pacient care nu a suferit niciodată de una este frecvent caracteristică, mai ales dacă atacurile de durere de cap (sau migrenele care sunt) sunt mai puternice dimineața și sunt asociate cu greață , vărsături și deficite neurologice. La pacienții care au suferit deja de cefalee, schimbarea fenomenologiei acestei tulburări sau creșterea frecvenței sau intensității atacurilor poate fi un semn al prezenței unei mase intracraniene. Convulsiile apar la aproximativ o treime din pacienții cu gliom, în special în cazurile de tumori de grad scăzut (vezi mai jos). Totuși, astfel de situații pot fi asociate cu orice tumoră SNC. Deficitul neurologic focal este legat de localizarea tumorii. Într-un procent între 15 și 20% dintre pacienții cu gliom există, de asemenea, o modificare a stării mentale. [4] [11]
Diagnostic pentru imagini
Prezența unei tumori cerebrale poate fi dezvăluită eficient prin tomografie computerizată (CT) și rezonanță magnetică nucleară (RMN). RMN este mai sensibil decât CT în identificarea leziunilor; cu toate acestea, nu este întotdeauna ușor de accesat pentru pacient și are unele contraindicații: nu poate fi efectuat persoanelor cu stimulatoare cardiace , proteze incompatibile cu câmpul magnetic , cleme metalice etc. CT rămâne metoda de alegere în detectarea calcificărilor în interiorul leziunilor sau eroziunilor osoase ale tecului sau bazei craniului. Utilizarea mediului de contrast, iodat în cazul CT, paramagnetic în cazul RMN ( gadoliniu ), permite achiziționarea de informații despre vascularizație și integritatea barierei hematoencefalice , o mai bună definiție a nodulului tumoral comparativ la edemul înconjurător și ne permite să facem ipoteze cu privire la gradul de malignitate. Examenul radiologic permite, de asemenea, să evalueze efectele mecanice (și consecințele modificări ale relațiilor structurilor creierului) derivate din prezența masei „străine”: hidrocefalie și hernii , ale căror efecte pot fi chiar letale. În cele din urmă, acest diagnostic, atunci când ne pregătim pentru operație, ne permite să specificăm locul leziunii și apropierea sau infiltrarea tumorii în zonele vitale ale creierului (așa-numitele zone „elocvente”). În acest scop, RMN este mai eficient decât CT datorită faptului că este capabil să furnizeze imagini tridimensionale. [12]

Instrumentele de diagnosticare a imaginii evidențiază fenomenul alterării din punct de vedere radiologic al țesutului neoplazic în comparație cu parenchimul cerebral normal (modificări ale densității electronice a materialelor în cazul CT și a intensității semnalului pentru RMN). La fel ca majoritatea țesuturilor patologice, tumorile se caracterizează și printr-o acumulare crescută de apă intracelulară. La CT apar hipodense, adică de densitate mai mică decât parenchimul creierului, la RMN apar hipointens în imaginile ponderate T1 și hiperintense în cele ponderate DP și T2. [13] [14]

La o radiografie, zona creierului sănătos nu ar trebui să prezinte nicio luminiscență specială. Prin urmare, este firesc să se acorde atenție porțiunilor celui mai mare semnal de contrast.
În tumoră, în general, cea mai mare pondere de îmbunătățire a contrastului se datorează barierei particulare hemato-tumorale, care permite trecerea iodului (CT) și a gadoliniului (RM) în spațiul interstițial extravascular intratumoral: astfel semnalul crește (densitatea sau intensitatea) tumorii. Cu toate acestea, trebuie acordată atenție faptului că îmbunătățirea contrastului nu delimitează cu certitudine neoplasmul de edem periwound: de fapt, în glioamele infiltrante maligne (cum ar fi, de exemplu, glioblastom și astrocitom anaplastic), constatarea anatomo-patologică arată țesutul neoplazic chiar și dincolo de edemul vasogen (care este cauzat de distrugerea barierei hematoencefalice de către tumoră), o stare clinică care nu este ușor de detectat prin imagistica diagnostic. [13] [14]
CT creierul prezintă de obicei o masă care poate (sau nu) să fie îmbunătățită de agentul de contrast. La CT, gliomii de grad scăzut (vezi mai jos) apar de obicei izodense față de parenchimul normal și, prin urmare, pot să nu prezinte îmbunătățirea contrastului. În mod similar, leziunile din fosa craniană posterioară sunt dificil de identificat pe CT. În consecință, rezultatele unei astfel de tomografii singure nu pot fi întotdeauna suficiente în scopuri diagnostice. [4]
Utilizarea RMN (mai sensibilă decât CT) este imperativă în cazurile îndoielnice pentru a confirma prezența unui neoplasm cerebral.
Pe imaginile MR ponderate T1, o tumoare intracraniană apare ca o leziune masivă care poate (sau nu) să devină mai luminiscentă după utilizarea agentului de contrast. Cu toate acestea, există întotdeauna o anomalie a semnalului în imaginile ponderate T2, indicând prezența unui neoplasm sau edem vasogen. De obicei, o luminescență mai mare ( îmbunătățirea contrastului ) indică o neoplasmă cu un grad mai mare de malignitate. Un inel de îmbunătățire este caracteristic glioblastomului (vezi imaginea), unde porțiunea luminiscentă corespunde părții vitale a tumorii maligne și regiunea mai întunecată (hipotensă în T1) corespunde zonei de necroză tisulară.
Atât rezonanța magnetică, cât și tomografia computerizată sunt de asemenea capabile să ofere informații fiziologice despre leziunile tumorale, dar, până în prezent, sensibilitatea și specificitatea acestor metode sunt prea mici pentru a constitui un protocol semeiotic definitiv. Un anumit diagnostic necesită utilizarea biopsiei sau rezecției chirurgicale, cu examinarea histologică a țesutului. [4]
Punerea în scenă
Majoritatea tumorilor intracraniene primare rămân localizate în craniu, deci nu sunt necesare proceduri sistemice de stadializare.
Tumorile neuroectodermice primare, meduloblastomul, tumorile celulelor germinale ale SNC și limfomul primar al SNC, pe de altă parte, se răspândesc frecvent prin spațiul subarahnoidian către leptomeningi . Prin urmare, RMN spinal este necesar pentru toți pacienții cu astfel de diagnostice. [4]
Tumori gliale (glioame)
Astrocitoame
Pentru gradarea (de malignitate) a astrocitoamelor, în literatura de specialitate au fost propuse de-a lungul timpului diferite sisteme (a se vedea în acest sens itemul Gradarea tumorilor sistemului nervos central ). Din 1993, sistemul de notare pe 4 niveluri propus de Organizația Mondială a Sănătății (OMS) a fost cel mai larg acceptat și răspândit. Se bazează pe patru caracteristici histologice: creșterea celularității, prezența mitozei , proliferarea endotelială , necroză . Conform sistemului de clasificare OMS (a se vedea Clasificarea tumorilor sistemului nervos central ), astrocitoamele de gradul I, cum ar fi astrocitomul pilocitar, sunt de obicei histologie benignă; astrocitoamele de gradul II (numite difuze ) au singura caracteristică histologică a celularității crescute și sunt neoplasme cu un grad scăzut de infiltrare; astrocitoamele de gradul III ( anaplazice ) prezintă, de asemenea, prezența semnificativă a mitozei; în astrocitoamele de gradul IV (glioblastom) se evidențiază și proliferarea endotelială și / sau necroza. [4]
Astrocitoame de grad scăzut

Tumorile circumscrise includ astrocitomul pilocitar (cu varianta pilomixoidă ), astrocitomul cu celule gigant subependimale și xanthastrocitomul pleomorf . Acestea sunt neoplasme mai puțin frecvente, cu histologie benignă și foarte adesea vindecabile numai cu o intervenție chirurgicală. Chiar dacă excizia este incompletă (din diverse motive), tumora poate rămâne indolentă sau poate fi tratată cu succes cu radioterapie. În cazurile rare în care tratamentul local eșuează, se utilizează tratamentul sistemic al chimioterapiei , a cărui amploare nu este însă găsită în unanimitate în literatura de specialitate; unele beneficii par să fi fost găsite în cazul copiilor cu combinație de carboplatină și vincristină . [4] [15]

La CT, astrocitoamele difuze de gradul II apar ca leziuni slab atenuante sau isointense. La RMN (metoda preferată) agentul de contrast poate să nu scoată în evidență imaginea acestor neoplasme, adică luminescența lor poate părea subțire și slabă.
Îmbunătățirea focală intensă poate indica zone cu anaplazie crescută. Ori de câte ori este posibil, se sugerează utilizarea biopsiei, pentru a obține probe din porțiunea luminiscentă: prognosticul, de fapt, este de obicei conectat la cea mai anaplazică parte a tumorii.
În majoritatea cazurilor, pacienții cu astrocitoame difuze sunt adulți tineri (a treia și a patra decadă de vârstă) care suferă de obicei convulsii. Condițiile pentru un prognostic favorabil includ vârsta tânără, dimensiunea tumorii mai mici de 5 cm și, atunci când este posibil, rezecția chirurgicală extinsă a tumorii. Supraviețuirea mediană este de aproximativ 5 ani. Recidivele tardive sunt relativ frecvente, astfel încât acești pacienți trebuie urmăriți timp de cel puțin 15 ani. [4]
În ciuda evoluției lor relativ indolente, majoritatea acestor astrocitoame progresează către leziuni caracterizate printr-o anaplazie mai mare, care nu sunt în mod normal vindecabile prin intervenții chirurgicale și radioterapie. Cu toate acestea, terapia pentru pacienții cu astrocitoame difuze de grad scăzut nu prezintă consens unanim în literatura de specialitate. Rolul rezecției „complete” este un subiect de dezbatere în contexte de specialitate. Rezultatele unor studii arată că excizia maximă a tumorii oferă cele mai bune rezultate. [16] De fapt, cazurile de rezecție completă implică adesea pacienți cu tumori mici și unilaterale, care nu implică structuri cerebrale critice. O abordare pragmatică, în general acceptabilă pentru majoritatea cazurilor, este aceea a unei excizii cât mai extinse posibil a țesutului neoplazic, evitând să provoace deficite neurologice semnificative. [4]

Radioterapia efectuată imediat după diagnostic s-a dovedit a prelungi timpul în care pacientul este lipsit de boli înainte de recurență, comparativ cu situația în care cursul radioterapiei este întârziat până la momentul progresiei. În prezent, însă, nu există o credință unanimă că radioterapia imediat după diagnostic îmbunătățește supraviețuirea generală a pacientului. [17]
Pentru pacienții cu simptome puține sau deloc sau cu convulsii care pot fi controlate cu medicamente anticonvulsivante , este acceptabilă întârzierea radioterapiei până când creșterea tumorii duce la o situație dificil de gestionat. Motivul radioterapiei întârziate rezidă adesea în dorința de a reduce riscul de afectare neurologică indusă chiar de radioterapie. Această motivație nu pare însă concludentă. [18]
Două studii clinice „randomizate” prospective nu au reușit în încercarea de a arăta un beneficiu mai mare în administrarea radioterapiei cu doze mari comparativ cu radioterapia cu doze mai mici. [19] [20] Tratamentul utilizat în mod obișnuit constă în administrarea totală între 45 și 54 Gy , efectuată cu fracții unice între 1,8 și 2,0 Gy. [4]
Rolul chimioterapiei adjuvante la pacienții cu astrocitoame de grad scăzut este încă în studiu. Rezultatele preliminare ale unui (faza 3) studiu clinic a comparat radioterapie singur cu radioterapie , urmat de procarbazină , lomustină și vincristină (PCV) chimioterapie arata ca combinatie de radio-chimioterapie ofera o perioada mai lunga de „supravietuirea fara boala. Dar nu a crescut supravietuirea globala. [21] Având în vedere toxicitatea asociată cu protocolul PCV, utilizarea temozolomidei este sugerată de multe părți, atât ca terapie inițială, cât și atunci când boala este recuperată. [22] [23] [24] [25] [26]
Astrocitom anaplastic

Pacienții cu astrocitom anaplastic prezintă de obicei convulsii, deficite neurologice focale, cefalee, modificări de personalitate. Vârsta medie la diagnostic este de aproximativ 45 de ani. RMN relevă, în general, prezența unei leziuni masive cu semnal de contrast crescut (îmbunătățire), deși există cazuri în care această îmbunătățire nu este evidențiată. Diagnosticul se stabilește cu examinarea histologică a materialului legat de leziune, luată prin biopsie sau rezecție chirurgicală. Prezența mitozei face posibilă distincția astrocitomului anaplastic de astrocitoamele de grad scăzut. Astrocitoamele anaplastice au o tendință ridicată de a se agrava în direcția anaplastică, prin urmare este necesar ca materialul care urmează să fie examinat să fie suficient pentru a permite distingerea tumorii de un glioblastom real. În special, un diagnostic histologic al astrocitomului anaplastic la un pacient care prezintă inelul clasic de îmbunătățire a glioblastomului pe rezonanță, sugerează că materialul adus la examinare nu este reprezentativ pentru leziune. [4] (Vezi și cazul prezentat în imaginile de mai sus: un glioblastom cu debut într-un astrocitom de grad scăzut .)

Indicii cu prognostic mai rău includ vârsta mai înaintată, starea fizică slabă, leziuni neurologice semnificative. În general, rezultatul terapeutic este mai bun cu o rezecție chirurgicală „completă”, dar nu este clar dacă acest rezultat mai bun trebuie asociat cu operația în sine sau cu scenariul clinic general care a permis o astfel de rezecție. [27]
Tratamentul standard la început prevede eliminarea maximă posibilă, încercând să nu crească posibilul deficit neurologic. Radioterapia este, de asemenea, standard în tratament, deoarece s-a demonstrat că prelungește perioada de supraviețuire. [4] [28] [29]
Rolul chimioterapiei este controversat. Unele studii clinice de fază 3 arată că pacienții pot beneficia de tratament de chimioterapie adăugat la radioterapie (comparativ cu radioterapia singură), în timp ce alte studii nu confirmă această situație. Utilizarea carmustinei singure sau a regimului PCV (procarbazină, lomustină și vincristină) a fost asociată cu creșterea supraviețuirii într-un studiu din 1999 [30] și într-o meta-analiză recentă. Acesta din urmă arată o creștere absolută de aproximativ 6% în supraviețuirea la 1 și 2 ani la pacienții supuși chimioterapiei. Iar supraviețuirea la doi ani este mai mare cu radioterapia plus chimioterapia (37%) decât cu radioterapia singură (31%). [31]
Spre deosebire de lucrările anterioare, totuși, un studiu clinic „randomizat” mare nu a verificat niciun beneficiu suplimentar în combinația de radioterapie plus PCV în comparație cu radioterapia singură. [32]

Chiar și în cazul utilizării temozolomidei (care este utilă în tratamentul recidivelor), un beneficiu suplimentar al chimioterapiei adăugat la radioterapie nu pare să fie clar stabilit. [4] [33]
Mediana de supraviețuire prezintă un interval de la 24 de luni la mai mult de 36 de luni. Lărgimea acestei game reflectă criteriile de selecție a pacientului. [4]
În caz de recidivă, utilizarea chimioterapiei nu ridică îndoieli: atât regimurile pe bază de nitrozure, cât și temozolomida au demonstrat eficacitate. Aprobarea acestui medicament din urmă de către Food and Drug Administration din SUA merge în acest sens. Răspunsul la temozolomidă este de 35% pentru pacienții care nu au primit chimioterapie anterioară și de 20% pentru pacienții care urmează următorul regim de chimioterapie (în special după nitrozuree). [34]
Glioblastom
Glioblastoamele sunt cele mai frecvente tumori ale gliei . Ele pot apărea de la zero sau pot rezulta dintr-un astrocitom difuz sau un astrocitom anaplastic .
Oligodendroglioame
Neoplasmele bazate pe oligodendroglia sunt relativ mai puțin frecvente, afectând mai puțin de aproximativ 5% din toate tumorile primare ale creierului [5] și nu mai mult de aproximativ 10-15% din glioame [35] (Vezi și Tabelele 1 și 2). Cu toate acestea, acestea sunt foarte importante pentru unicitatea sensibilității la chimioterapie.
Aceste tumori sunt împărțite în leziuni de „grad scăzut” și leziuni anaplastice. Oligodendrogliomul anaplastic se caracterizează prin celularitate ridicată, polimorfism nuclear, mitoză frecventă, proliferare abundentă endotelială și necroză. [4]
Aproximativ jumătate din oligodendroglioamele se caracterizează prin pierderea heterozigozității cromozomilor 1p și 19q, o caracteristică tipică (patognomică) pentru diagnostic. (Recent s-a demonstrat că o astfel de pierdere de heterozigoză este secundară translocației pericentromerice dezechilibrate. [36] [37] )
Majoritatea oligodendroglioamelor apar ca o tumoare de grad scăzut. Glioamele mixte, precum oligoastrocitomul și oligoastrocitomul anaplastic, conțin atât componente oligodendrogliale, cât și componente astrocitice.
Oligodendrogliom / oligoastrocitom de grad scăzut

Supraviețuirea medie pentru pacienții cu oligodendrogliom "pur" este de aproximativ 10 ani; cel al pacienților cu oligoastrocitom este de aproximativ 8 ani (deci intermediar între cel al unui oligodendrogliom pur și cel al unui astrocitom pur). Ștergerea (sau translocarea) perechii 1p / 19q din tumoră este asociată cu o supraviețuire mai lungă. [37]
L'età media alla diagnosi è di 35 anni. La sintomatologia tipica annovera crisi epilettiche, ma possono pure segnalarsi deficit neurologici focali, modifiche della personalità ovvero gli altri sintomi di pressione endocranica (cefalea, vomito, ecc.). Questi tumori non sono normalmente visibili alla TC, quindi la metodica di elezione per la diagnostica per immagini risulta la risonanza magnetica. Alla RM sono visibili come aumentata intensità di segnale nelle immagini T2-pesate. Alle immagini T1-pesate il segnale può risultare attenuato e il contrast enhancement captato solo occasionalmente. Può essere o no presente la mancanza di segnale da calcificazione. [4]

Tali neoplasie sono più indolenti delle corrispondenti astrocitarie e, come per gli astrocitomi di basso grado, anche per questi tumori non c'è accordo nella letteratura per quanto riguarda il trattamento ottimale. È stata avanzata l'ipotesi che il beneficio di una escissione il più completa possibile sia rilevante, ma è da tener presente che spesso si tratta di casi di tumori di dimensioni non cospicue in zone non vitali del cervello. I primi risultati di un test clinico europeo non ha mostrato benefici, in termini di sopravvivenza, della radioterapia fatta immediatamente dopo l'intervento chirurgico rispetto a quella ritardata sino alla comparsa di una sintomatologia che non si riesca a controllare farmacologicamente, benché nei casi di radioterapia immediata è stato evidenziato un maggior lasso di tempo di assenza di sintomi prima della nuova progressione del tumore. [17]

Due altri test clinici non hanno evidenziato benefici in una radioterapia a più alte dosi rispetto a una a dosi “intermedie”. [19] [20]
Dati provenienti da diversi studi [38] [39] [40] indicano che una terapia iniziale con temozolomide o PCV può rimpicciolire un oligodendroglioma o un oligoastrocitoma in una percentuale che varia dal 31% al 61% dei casi. Ma è ancora da stabilire se questa “risposta” alla chemioterapia migliori la sopravvivenza globale del paziente ovvero aumenti solo la durata del periodo di scarsa sintomatologia prima della successiva fase di progressione recidiva verso situazioni più complesse da gestire.
Un test clinico [21] progettato per confrontare la radioterapia da sola con la radioterapia seguita da PCV verificò un periodo medio di sopravvivenza senza malattia superiore nel gruppo col PCV, ma nessuna differenza sostanziale in termini di sopravvivenza globale tra i due gruppi. Alla ricorrenza ad entrambi i gruppi veniva somministrato il PCV, significando con ciò che si ottiene lo stesso risultato indipendentemente da quando si comincia la chemioterapia (prima o dopo la ricorrenza).
Anche con la temozolomide si ottiene una riduzione dell'azione tumorale, ed essendo meno tossica del PCV, viene da più parti preferito il suo uso. [4]
Per riassumere, il trattamento iniziale (cioè non al momento della recidiva) prevede il controllo dei sintomi con solo anticonvulsivi , la radioterapia da sola, la chemioterapia da sola o la combinazione di radioterapia più chemioterapia. Alla recidiva, chirurgia, radioterapia e chemioterapia svolgono tutte un ruolo importante. Una seconda resezione (o una prima, se non era mai stata fatta) può ridurre i sintomi. Se la radioterapia non era stata fatta nel trattamento iniziale, è probabile che risulti efficace contro la recidiva. Una risposta alla temozolomide si registra in circa il 50% dei pazienti che presentino ricorrenza dopo la radioterapia. [41] [42]
Oligodendroglioma/Oligoastrocitoma anaplastico



I tumori oligodendrogliali anaplastici si presentano con i sintomi tipici che derivano dall'effetto massa e con crisi epilettiche. Nonostante la loro chemiosensibilità, la mediana di sopravvivenza va da 3 a 5 anni soltanto. Il trattamento prevede l'escissione più ampia possibile, seguita da radioterapia.
Per quel che riguarda la chemioterapia si osservi quanto segue: due recenti test clinici di fase 3, l'uno condotto negli Stati Uniti [43] e l'altro in Europa [44] hanno messo a confronto la radioterapia da sola con la coppia radioterapia più PCV. Nello studio americano i pazienti di un gruppo ricevevano 4 cicli di PCV prima della radioterapia (l'altro gruppo la sola radioterapia). Benché il periodo di vita senza sintomi rilevanti sia risultato più lungo nel gruppo col PCV, la sopravvivenza globale è risultata la stessa nei due gruppi. I pazienti con delezione in 1p/19q ottenevano i risultati migliori; ma anche i pazienti senza delezione in 1p/19q miglioravano la loro performance col PCV.
Nello studio europeo i pazienti ricevevano 6 cicli di PCV dopo la radioterapia, ma i risultati sono stati quasi identici a quelli dello studio americano. La sopravvivenza libera da malattia era migliore nel gruppo del PVC, ma ancora una volta, la sopravvivenza globale non differiva nei due gruppi. I pazienti con delezione in 1p e 19q avevano una sopravvivenza di qualità superiore, indipendentemente dal gruppo di appartenenza. Non è stata osservata differenza significativa nella sopravvivenza globale nei due gruppi relativamente a delezione o meno in 1p/19q.
I risultati di questi due studi combinati mostrano che la chemioterapia migliora la sopravvivenza libera da malattia ma che il trattamento di salvataggio alla recidiva fornisce come esito una sopravvivenza globale equivalente. Cosa importante, entrambi i test clinici confermano il valore prognostico della coppia 1p/19q, ma non mostrano in maniera definitiva che solo i pazienti con delezione in 1p e 19q beneficiano della chemioterapia.
Studi clinici prospettici hanno mostrato che, approssimativamente, dal 50% al 70% dei pazienti con oligodendroglioma anaplastico ricorrente dopo la radioterapia risponde alla chemioterapia con PCV o temozolomide. [45] Benché non vi sia riscontro che la sequenza di temozolomide e PCV abbia un'efficacia superiore, l'assenza di mielosoppressione [46] cumulativa con la temozolomide suggerisce il suo uso all'inizio del trattamento della recidiva. [4]
Ependimoma

L'ependimoma è una neoplasia che si sviluppa dalle cellule ependimali , che rivestono i ventricoli , il plesso corioideo , il filum terminale e il canale centrale del midollo spinale . Cellule ependimali sono pure presenti nel parenchima cerebrale quale risultato di migrazione da aree periventricolari alla corteccia cerebrale , durante lo stadio embrionale . [47] [48]
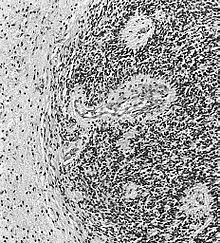
Questo tipo di tumore può comparire ad ogni età, ma presenta due picchi caratteristici, uno da zero a 10 anni ed un altro tra i 40 ei 50 anni. Le lesioni intracraniche (di solito nella fossa cranica posteriore) sono più comuni nella prima fascia di età, quelle spinali nella seconda. [47]
Come si vede dalle Tabelle 1 e 2, si tratta di tumori abbastanza rari, sia in assoluto tra le neoplasie del sistema nervoso (2,3%), sia tra i gliomi (5,6%).
Si distinguono in lesioni di basso grado (I e II della scala WHO) e lesioni anaplastiche (III della scala WHO). In particolare il subependimoma e l' ependimoma mixopapillare sono di grado I,
l' ependimoma è di grado II, l' ependimoma anaplastico è di grado III. (Vedi la Classificazione dei tumori del sistema nervoso centrale .)
Gli ependimomi di basso grado nella spina dorsale resecabili vengono trattati con la sola chirurgia.
Mentre il ruolo della radioterapia postchirurgica per gli ependimomi intracranici di basso grado rimane controversa, i tumori anaplastici o quelli di basso grado non completamente escissi sono normalmente trattati con radioterapia.
Studi clinici hanno mostrato che gli ependimomi rispondono ai regimi chemioterapici, soprattutto a quelli basati sul platino . [49] Dallo studio appena citato si evince infatti che la chemioterapia basata su platino fornisce il 67% delle risposte, mentre i regimi basati su nitrosurea hanno una risposta del 25%.
Per quanto riguarda la prognosi, gli ependimomi di grado II hanno una sopravvivenza libera da malattia a 6 anni del 66% ed una sopravvivenza globale dell'87%; per gli ependimomi anaplastici questi valori scendono rispettivamente al 29% e al 37%. [47]
Tumori non gliali
Medulloblastoma
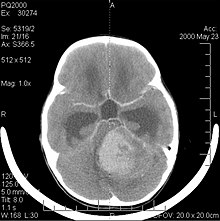
Il medulloblastoma, come altre neoplasie embrionali quali il tumore cerebrale neuroectodermico primitivo o il neuroblastoma cerebrale , è un tumore maligno del sistema nervoso centrale molto raro nella popolazione adulta (oltre 21 anni). È il tumore cerebrale maligno più frequente nell' infanzia , anche i giovani ne sono però a rischio. Il picco d'incidenza si verifica nei bambini di età tra i 2 ei 7 anni. Questo tumore è tipico della fossa cranica posteriore , ove si localizza in entrambi gli emisferi del cervelletto ovvero nel verme cerebellare ed essendo invasivo ed a rapida crescita usualmente diffonde ad altre parti del sistema nervoso centrale attraverso il liquor: può infiltrare il pavimento del vicino quarto ventricolo ed estendersi nella sua cavità, può anche passare nelle meningi . Più raramente, può dare metastasi extracraniche. La sintomatologia al presentarsi della neoplasia includono perdita di equilibrio, mancanza di coordinazione, diplopia , disartria e, a causa del coinvolgimento del quarto ventricolo (per il quale è comune un idrocefalo ostruttivo), i segni dell' idrocefalo , includenti cefalea, nausea, vomito, andatura instabile. [4]
La risonanza magnetica usualmente rivela una lesione massiva a significativo contrast enhancement coinvolgente il cervelletto. Come sopra si diceva, il medulloblastoma ha un'alta propensione ad infiltrare focalmente le leptomeningi , così come a propagare attraverso lo spazio subaracnoideo per coinvolgere i ventricoli, la convessità cerebrale, le superfici leptomeningee spinali. Di conseguenza risulta necessario sottoporre a risonanza l'intero asse cranio-spinale. [4]

È affidato alla chirurgia il compito di rimuovere quanto più è possibile della massa rappresentata dalla lesione, infatti residui tumorali postchirurgici sono causa di una prognosi peggiore. Foriera di prognosi non favorevole è anche la presenza di cellule neoplastiche nel liquido cerebrospinale ovvero la rilevazione alla risonanza di metastasi leptomeningee.
La chirurgia da sola di solito non è sufficiente come terapia, tuttavia lo può risultare in certi casi la successiva radioterapia all'asse cranio-spinale, con focalizzazione sul sito del tumore primario.
La chemioterapia dopo la radioterapia, inoltre, aumenta il tasso di guarigione. Si usano farmaci a base di platino (cisplatino o carboplatino), l'etoposide, e un agente alchilante (ciclofosfamide o lomustina) insieme alla vincristina. [4] Con un appropriato trattamento i casi di lunga sopravvivenza (superiore a 3 anni), per i pazienti di medulloblastoma, vanno dal 60% all'80%. [50]
Meningioma

I meningiomi sono i tumori intracranici più diffusi (Vedi Tabella 1). Sono solitamente benigni ed originano dall' aracnoide , membrana che ricopre il cervello e il midollo spinale. L'incidenza di questo tipo di neoplasie è di circa 2 casi all'anno ogni 100.000 abitanti. Sono più comuni nelle donne, nella sesta e settima decade di vita. La loro frequenza è maggiore per i pazienti con neurofibromatosi di tipo 2 . La perdita del cromosoma 22 è caratteristica dei meningiomi, benché ancora non sia chiaro il significato prognostico di questa scoperta.
Nonostante che questa lesione abbia espressi recettori per androgeni , estrogeni , progesterone e somatostatina , le terapie dirette all'utilizzo di questi recettori non hanno ancora mostrato efficacia. [4]
I pazienti con meningioma possono presentare la sintomatologia tipica di una lesione massiva nella scatola cranica, incluse crisi epilettiche e deficit neurologici focali. [51] [52]

Il meningioma, che può essere anche asintomatico, risulta talvolta scoperto incidentalmente da TC o risonanza magnetica, effettuate per altre ragioni. Questo tumore alla risonanza ha un aspetto caratteristico, che consiste, di norma, in un contrast enhancement uniforme lungo la dura, con netta separazione dal parenchima cerebrale. Altra caratteristica (benché non presente in tutti i casi) è la cosiddetta “coda durale”, rappresentato da enhancement che si estende oltre la lesione, ad indicare il punto di ancoraggio nella dura . [53]
Solita è la presenza di edema peritumorale, conseguenza del fattore di crescita vascolare endoteliale secreto dalle cellule neoplastiche, che influenza a sua volta l'effetto massa locale.
Molti meningiomi scoperti incidentalmente non necessitano di trattamento al momento della diagnosi iniziale. Per i pazienti con mengiomi asintomatici può risultare appropriato e sufficiente tenere la lesione sotto osservazione. L'evidenza epidemiologica suggerisce che i due terzi di questi pazienti non avrà sintomatologia straordinaria. [54]
Se si riscontra nel paziente un significativo effetto massa, che vi siano o no dei sintomi, il trattamento di elezione è normalmente la resezione completa. L'escissione è spesso realizzabile se il meningioma è situato sulla convessità cerebrale, il solco olfattivo , il seno sagittale superiore o la fossa posteriore . La resezione può risultare molto più difficoltosa se il tumore si presenta in altri siti, quali le regioni sfenoidale, parasagittale, orbitale, tentoriale o del clivus. In tali circostanze per il controllo del tumore risultano oltremodo utili la radioterapia classica o la radiochirurgia stereotassica. [4]
In uno studio della Mayo Clinic , che confrontò la percentuale di controllo del tumore dopo resezione chirurgica e con radiochirurgia, in pazienti con meningioma intracranico di dimensioni medio-piccole e senza sintomi da effetto massa, [55] la radiochirurgia risultò ottenere un migliore controllo del tumore (98% contro 88%) e con minori complicanze (10% contro 22%) rispetto alla escissione chirurgica.
La chirurgia stereotassica è normalmente riservata alle lesioni più piccole (cioè inferiori a 3–4 cm), laddove per lesioni più grandi ovvero prossime a strutture critiche, come i nervi ottici, si usa la radioterapia frazionata.
Per quanto riguarda la chemioterapia, ad oggi nessun intervento farmacologico ha mostrato efficacia antitumorale riproducibile.
Raramente i meningiomi presentano caratteristiche istologiche atipiche o di franca malignità. In questi casi però risultano altamente aggressivi. L'approccio a tali tumori è identico a quello visto per i tumori benigni, con la differenza che la radioterapia postchirurgica diventa usuale e non episodica. [4]
Linfoma primario del SNC

I linfomi primari del sistema nervoso centrale (cioè che sorgono nel SNC in assenza di linfoma al di fuori del SNC al momento della diagnosi) [56] costituiscono approssimativamente dal 2% al 3% di tutti i tumori cerebrali dei pazienti con un normale sistema immunitario (Cfr. Tabella 1).
La neoplasia è più comune nei maschi dai 55 ai 60 anni; quasi metà di tutti i linfomi si hanno in pazienti che hanno più di 60 anni e circa un quarto in pazienti con più di 70. L'incidenza sembra che stia aumentando, anche se non è chiaro se tale aumento sia reale o rifletta un'alterazione di rilevazione.
Esposti a un maggior rischio di linfoma del SNC sono sicuramente i pazienti con un sistema immunitario compromesso, quindi coloro che hanno subito un trapianto d'organi, quelli che hanno un' immunodeficienza congenita o una malattia autoimmune o che sono infetti dal virus dell' AIDS . I linfomi cerebrali associati al virus dell'immunodeficienza sono collegati con il virus di Epstein-Barr , in particolare nei pazienti con un conteggio di linfociti CD4 [57] inferiore a 500 cellule per millimetro cubo (di sangue).
La maggior parte dei linfomi del SNC sono del tipo a grandi cellule B . [4]
I pazienti presentano una varietà di sintomi caratteristici di lesione massiva focale o multifocale. La risonanza mostra di solito tumori, con contrast enhancement omogeneo, all'interno della materia bianca periventricolare profonda. Multifocalità ed enhancement disomogeneo sono tipici in pazienti con sistema immunitario compromesso.
Estremamente importante è l'analisi del linfoma del SNC nella diagnosi differenziale delle neoplasie cerebrali. Si tenga conto che la somministrazione di corticosteroidi può dare come risultato la completa scomparsa dell' enhancement lesionale, rendendo difficoltosa la diagnosi. Di conseguenza, quando si consideri un linfoma del SNC in diagnosi differenziale occorre evitare i corticosteroidi, a meno che l'effetto massa non stia causando un serio ed immediato problema al paziente.
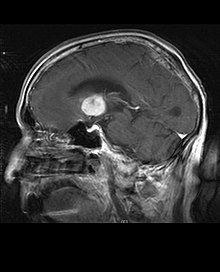
Criticamente importante è l'ottenimento di un campione bioptico della sospetta lesione, in quanto molte malattie del SNC, maligne e non, possono apparire un linfoma.
Diversamente dai linfomi sistemici “a cellule B grandi”, per i quali sia la chemioterapia che la radioterapia sono efficaci e il trattamento di lesioni localizzate è curativo, il linfoma del sistema nervoso centrale tipicamente risponde alla terapia iniziale ma poi recidiva. Come per il linfoma sistemico, il ruolo della chirurgia è ristretto soprattutto all'ottenimento di appropriati campioni di tessuto per la diagnosi. [4]
La radioterapia dell'intero cervello (panencefalica) era una volta la strada maestra del trattamento. Sfortunatamente, anche con lesioni localizzate, la mediana di sopravvivenza con la sola radioterapia è di circa 1 anno. La recidiva interessa di solito il sito della precedente lesione oltre ad altre regioni. Interessanti sono le risposte con la chemioterapia.
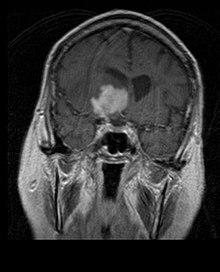
Hanno mostrato di fornire una migliore sopravvivenza globale, rispetto alla sola radioterapia, studi clinici nei quali è stato usato del metotrexato ad alte dosi, da solo, come primo trattamento e rinviando la radioterapia al momento della recidiva/progressione; [58] il mix metotrexato, vincristina, procarbazina, metotrexato intratecale, radioterapia panencefalica, citarabina; [59] ovvero la chemioterapia intraarteriosa (metotrexato per via intraarteriosa, ciclofosfamide ed etoposide per via intravenosa), dopo la modifica della barriera emato-encefalica con mannitolo . [60]
Nei regimi con metotrexato la mediana di sopravvivenza è risultata di molto superiore a quella associata alla radioterapia da sola (intervallo da 24 a 40 mesi). [58] In alcuni casi la radioterapia è usata solo alla recidiva, in caso di regressione iniziale ottenuta con la chemioterapia; sono riportati casi di lunga sopravvivenza anche senza l'uso di radioterapia. [58] [59] [60]
Per la natura diffusa del linfoma del SNC, normale è l'affidamento alla radioterapia panencefalica , la quale però porta con sé un alto rischio di demenza , secondaria a leucoencefalopatia . Questo rischio potrebbe essere ridotto con lo sviluppo di strategie di controllo efficace del tumore che evitino la radioterapia panencefalica.
La terapia iniziale per i pazienti con sistema immunitario compromesso è la riduzione delle cause di immunosoppressione. La prognosi per questi pazienti è normalmente peggiore di quella per i pazienti con alla diagnosi un sistema immunitario normale. A causa di infezioni concomitanti il tumore e una condizione fisica di solito non ottimale, in questi pazienti immunodepressi spesso la chemioterapia non può essere somministrata.
Come per le altre neoplasie cerebrali, la risposta ai trattamenti è correlata all'età ed alla condizione fisica. [4]
Tumori del sistema nervoso centrale – Tumori metastatici
Metastasi cerebrali

Le metastasi al cervello sono le neoplasie intracraniche più comuni negli adulti 10 volte più frequenti dei precedenti tumori cerebrali primari . Si verificano nel 20-40% degli adulti con cancro e sono associate soprattutto con il carcinoma polmonare e mammario e col melanoma.
Queste lesioni sono il risultato della propagazione di cellule cancerose attraverso il flusso sanguigno e sono massimamente presenti alla connessione della materia grigia con quella bianca, dove il calibro dei vasi sanguigni cambia, intrappolando così gli emboli cancerosi.
L'80% delle lesioni si verificano negli emisferi cerebrali, il 15% nel cervelletto e il 5% nel tronco encefalico. Approssimativamente l'80% dei pazienti hanno una storia di cancro sistemico e il 70% presentano metastasi cerebrali multiple. [4]
Sostanziali passi avanti si sono fatti negli ultimi tempi nella diagnosi e nel trattamento di queste lesioni, migliorando sopravvivenza e controllo della sintomatologia.
Segni e sintomi alla presentazione sono simili a quelli delle altre lesioni massive nel cervello.
Lo strumento diagnostico di elezione è la risonanza con mezzo di contrasto. Tuttavia, nei pazienti di cancro, non tutte le lesioni cerebrali sono metastasi. In uno studio clinico prospettico su pazienti con cancro sistemico sospetti di avere una metastasi cerebrale singola, l'11% dei campioni di tessuto mostrò invece un tumore cerebrale primario o un'infiammazione ovvero un'infezione. [61]

Due studi prospettici randomizzati [61] [62] hanno mostrato che chirurgia più radioterapia panencefalica producono migliori risultati della sola chirurgia, in pazienti selezionati. Cioè in buone condizioni fisiche, con una lesione sistemica stabile o limitata e con una metastasi cerebrale singola chirurgicamente accessibile. Chirurgia più radioterapia danno come risultato un numero inferiore di decessi per cause neurologiche rispetto alla chirurgia da sola. [62] Tuttavia l'aggiunta di radioterapia panencefalica non migliora la sopravvivenza globale rispetto alla sola chirurgia. [62]

Per le lesioni difficilmente trattabili chirurgicamente può risultare efficace la radiochirurgia stereotassica. Due studi prospettici randomizzati [63] [64] hanno verificato che pazienti selezionati con un numero limitato di metastasi cerebrali hanno avuto maggior giovamento quando trattati con radiochirurgia più radioterapia panencefalica che con trattamento di sola radioterapia a tutto il cervello.
Un test clinico prospettico [65] ha raffrontato l'efficacia di chirurgia e radiochirurgia, randomizzando i pazienti con singola piccola metastasi in un primo gruppo con chirurgia seguita da radioterapia panencefalica e un secondo gruppo con sola radiochirurgia. Non è stata trovata differenza significativa nei risultati. Analisi retrospettive hanno riportato risultati in conflitto. [66] [67]
Ricapitolando, la letteratura mostra risultati equivalenti per chirurgia e radiochirurgia. Quest'ultima sembra più conveniente, efficace e sicura per lesioni piccole o in regioni inaccessibili alla chirurgia. La radiochirurgia offre un'alternativa ragionevole a pazienti che non sono candidabili alla chirurgia per ragioni mediche. Tuttavia la chirurgia è chiaramente la modalità ottimale per ottenere tessuto per la diagnosi e per l'escissione di lesioni che causano effetto massa. Quindi, radiochirurgia e chirurgia andrebbero meglio considerate come due metodiche complementari ma differenti, da usare ciascuna a seconda della diversa situazione del paziente. [68]
Prima di concludere questa parte, si osservi che, nella realtà, quasi il 50% dei pazienti con 1 o 2 metastasi cerebrali non sono candidabili per l'asportazione chirurgica a causa dell'inaccessibilità delle lesioni, l'estensione della malattia sistemica ovvero per altri fattori di complicazione. A questi pazienti, e ad altri con metastasi multiple, normalmente si offre come trattamento standard la radioterapia panencefalica. Con tale terapia in effetti sino a quasi il 50% di essi ottiene un miglioramento dei sintomi neurologici e il 50-70% mostra una risposta obiettiva. [69] [70] [71]
La chemioterapia raramente ha il ruolo di terapia primaria nel caso di metastasi cerebrali. Molte neoplasie che metastasizzano al cervello (per es., il carcinoma polmonare non a piccole cellule, neoplasie in cui il sito di origine primario è sconosciuto o il melanoma) sono insensibili alla terapia farmacologica o risultano già pesantemente trattate con agenti che si riteneva potenzialmente efficaci. [4]
Per la maggior parte dei pazienti con metastasi cerebrali la mediana di sopravvivenza è di soli 4-6 mesi, dopo la radioterapia panencefalica. Tuttavia alcuni pazienti (con età inferiore a 60 anni, lesione singola e malattia sistemica sotto controllo) possono raggiungere una sopravvivenza maggiore, per il fatto che sono in grado di essere sottoposti a un approccio terapeutico più aggressivo. Ad esempio, una parte di questi pazienti riescono ad affrontare un'altra operazione chirurgica o la radiochirurgia stereotassica. Con trattamento aggressivo la mediana di sopravvivenza arriva a 40 settimane e più. [72]
Metastasi leptomeningee

Il coinvolgimento delle leptomeningi avviene in circa il 5% dei pazienti di cancro e viene rilevato più frequentemente, mano in mano che le metodiche diagnostiche migliorano ed i pazienti vivono più a lungo. Le neoplasie di origine più comuni sono il melanoma ei carcinomi mammario e polmonare. Il cancro raggiunge le leptomeningi come risultato della propagazione di cellule cancerose attraverso il flusso sanguigno. Le cellule maligne risultano in genere disseminate per tutto il nevrasse dal flusso del liquido cerebrospinale.
I segni ei sintomi sono riferibili ad una o più delle seguenti situazioni: danno locale ai nervi che viaggiano attraverso il fluido spinale (paralisi dei nervi cranici, debolezza motoria con comparsa di dolori radicolari , parestesie , fitte ); invasione diretta del cervello o dei tessuti spinali; interruzione dei vasi sanguigni diretti a quei tessuti (deficit neurologici focali o attacchi epilettici); ostruzione del normale flusso del liquido cerebrospinale (cefalea ed aumento della pressione endocranica); interferenza con il normale funzionamento del cervello ( encefalopatia ); ovvero infiltrazione perivascolare da parte di cellule tumorali, con conseguente ischemia locale e sintomi da colpo apoplettico.
La diagnosi si effettua con l'esame del liquido cerebrospinale e/o la risonanza magnetica del cervello e del midollo spinale. Lo studio del liquor rivela la presenza di cellule maligne nel 50% dei pazienti; tuttavia in almeno il 10% dei malati con sospetto coinvolgimento leptomeningeo l'esame citologico rimane persistentemente negativo. L'aumento del numero di punture lombari (fino a 6) e del volume di liquido rimosso (10 ml per puntura) incrementa la possibilità di diagnosi positiva. Nel liquido cefalorachidiano la concentrazione di proteine è normalmente elevata, quella di glucosio può essere bassa, con presenza di pleocitosi . Lo studio radiologico può evidenziare idrocefalo in assenza di lesione massiva o enhancement diffuso delle leptomeningi.
Senza terapia la mediana di sopravvivenza è di 4-6 settimane, con decesso dovuto a progressivo deterioramento neurologico. Spesso le metastasi leptomeningee sono una manifestazione dello stadio finale della malattia principale e la terapia sintomatica può essere la soluzione più appropriata. Corticosteroidi ed analgesici offrono un temporaneo alleviamento. Ai pazienti con malattia sistemica minimale ed accettabile condizione fisica generale può essere offerto un trattamento per attenuare i sintomi e prolungare la sopravvivenza.
La sopravvivenza mediana può essere aumentata da 3 a 6 mesi con radioterapia ai siti sintomatici e delle aree malate più voluminose individuate con lelastre, e con terapia intratecale con metotrexato , citarabina e tiotepa (effettuata con puntura lombare o catetere Ommaya ).
Benché la chemioterapia riesca a prolungare significativamente la sopravvivenza di pazienti con malattia ematologica, tipo leucemia o anche linfoma, ottenere un beneficio attraverso il liquido cerebrospinale quando si ha a che fare con tumori solidi risulta perlomeno dubbio. In tali circostanze il decesso avviene per malattia sistemica avanzata.
La maggior complicanza della terapia intratecale a base di metotrexato è rappresentata da una leucoencefalopatia necrotizzante [73] che può svilupparsi dopo mesi di terapia in quei pochi pazienti che giovano di una sopravvivenza prolungata. Questo effetto tossico devastante è comune soprattutto nei pazienti sottoposti a radioterapia precedente o contemporaneamente alla terapia intratecale con metotrexato. [4]
Note
- ^ ( EN ) Farlex Medical Dictionary , su medical-dictionary.thefreedictionary.com . URL consultato il 24 aprile 2011 .
- ^ ( EN ) Mondofacto Online Medical Dictionary , su mondofacto.com . URL consultato il 24 aprile 2011 (archiviato dall' url originale il 7 novembre 2011) .
- ^ ( EN ) Levin VA, Neuro-oncology: an overview ( PDF ), in Arch Neurol , vol. 56, n. 4, aprile 1999, pp. 401-4, PMID 10199326 .
- ^ a b c d e f g h i j k l m n o p q r s t u v w x y z aa ab ac ad ae af ( EN ) Buckner JC, Brown PD, O'Neill BP, Meyer FB, Wetmore CJ, Uhm JH, Central nervous system tumors ( PDF ) [ collegamento interrotto ] , in Mayo Clin Proc , vol. 82, n. 10, ottobre 2007, pp. 1271-86, DOI : 10.4065/82.10.1271 , PMID 17908533 .
- ^ a b ( EN ) DeAngelis LM, Gutin PH; Leibel SA; Posner JB, Intracranial Tumors: Diagnosis and Treatment , Informa Healthcare, 2002, ISBN 1-901865-37-1 .
- ^ a b c d ( EN ) Central Brain Tumor Registry of the United States, Primary brain tumors in the United States. Statistical report 1998-2002 ( PDF ), in Department of Health and Human Services, Centers for Disease Control and Prevention (CDC), National Program of Cancer Registries (NPCR) , 2005. URL consultato il 24 aprile 2011 (archiviato dall' url originale il 2 marzo 2011) .
Come citato in: Buckner JC, Brown PD, O'Neill BP, Meyer FB, Wetmore CJ, Uhm JH, Central nervous system tumors ( PDF ) [ collegamento interrotto ] , in Mayo Clin Proc , vol. 82, n. 10, ottobre 2007, pp. 1271-86, DOI : 10.4065/82.10.1271 , PMID 17908533 . - ^ ( EN ) Jemal A, Siegal R, Ward E, Murray T, Xu J, Thun MJ, Cancer statistics, 2007 ( PDF ) [ collegamento interrotto ] , in CA Cancer J Clin , vol. 57, n. 1, gennaio-febbraio 2007, pp. 43-66, DOI : 10.3322/canjclin.57.1.43 , PMID 17237035 .
- ^ Il glioblastoma è il tumore più comune che colpisce il cervello. Il meningioma è meno raro del glioblastoma, ma per quanto detto all' inizio della voce a rigor di termini non è un tumore cerebrale .
- ^ ( EN ) Garrè ML, Cama A, Bagnasco F, Morana G, Giangaspero F, Brisigotti M, Gambini C, Forni M, Rossi A, Haupt R, Nozza P, Barra S, Piatelli G, Viglizzo G, Capra V, Bruno W, Pastorino L, Massimino M, Tumolo M, Fidani P, Dallorso S, Schumacher RF, Milanaccio C, Pietsch T, Medulloblastoma Variants: Age-Dependent Occurrence and Relation to Gorlin Syndrome--A New Clinical Perspective ( PDF ), in Clin Cancer Res , vol. 15, n. 7, 1º aprile 2009, pp. 2463-71, DOI : 10.1158/1078-0432.CCR-08-2023 , PMID 19276247 .
- ^ ( EN ) Wrensch M, Minn Y, Chew T, Bondy M, Berger MS, Epidemiology of primary brain tumors: current concepts and review of the literature , in Neuro Oncol , vol. 4, n. 4, ottobre 2002, pp. 278-99, DOI : 10.1093/neuonc/4.4.278 , PMID 12356358 .
- ^ ( EN ) Posner JB, Neurologic Complications of Cancer , 1ª ed., Philadelphia, PA, Oxford University Press, 1995, ISBN 0-8036-0006-2 .
- ^ Bruzzone MG, Farina L, Imaging dei gliomi cerebrali , su biometis.unimi.it . URL consultato il 24 aprile 2011 (archiviato dall' url originale l'8 maggio 2008) .
- ^ a b Macchi G., Minciacchi D; Gainotti G, Malattie del sistema nervoso , 2ª ed., PICCIN, 2005 [1981] , ISBN 88-299-1739-7 .
- ^ a b Biagini C, Gavelli G, Radiobiologia e radioprotezione , PICCIN, 1999, ISBN 88-299-1463-0 .
- ^ ( EN ) Packer RJ, Ater J, Allen J, Phillips P, Geyer R, Nicholson HS, Jakacki R, Kurczynski E, Needle M, Finlay J, Reaman G, Boyett JM, Carboplatin and vincristine chemotherapy for children with newly diagnosed progressive low-grade gliomas , in J Neurosurg , vol. 86, n. 5, maggio 1997, pp. 747-54, DOI : 10.3171/jns.1997.86.5.0747 , PMID 9126887 .
- ^ ( EN ) Keles GE, Lamborn KR, Berger MS, Low-grade hemispheric gliomas in adults: a critical review of extent of resection as a factor influencing outcome , in J Neurosurg , vol. 95, n. 5, novembre 2001, pp. 735-45, DOI : 10.3171/jns.2001.95.5.0735 , PMID 11702861 .
- ^ a b ( EN ) van den Bent MJ, Afra D, de Witte O, Ben Hassel M, Schraub S, Hoang-Xuan K, Malmström PO, Collette L, Piérart M, Mirimanoff R, Karim AB; EORTC Radiotherapy and Brain Tumor Groups and the UK Medical Research Council, Long-term efficacy of early versus delayed radiotherapy for low-grade astrocytoma and oligodendroglioma in adults: the EORTC 22845 randomised trial , in Lancet , vol. 366, n. 9490, settembre 2005, pp. 985-90, DOI : 10.1016/S0140-6736(05)67070-5 , PMID 16168780 . Erratum: ( EN ) Department of Error, Erratum , in Lancet , vol. 367, n. 9525, 3 giugno 2006, p. 1818, DOI : 10.1016/S0140-6736(06)68803-X .
- ^ ( EN ) Klein M, Heimans JJ, Aaronson NK, van der Ploeg HM, Grit J, Muller M, Postma TJ, Mooij JJ, Boerman RH, Beute GN, Ossenkoppele GJ, van Imhoff GW, Dekker AW, Jolles J, Slotman BJ, Struikmans H, Taphoorn MJ, Effect of radiotherapy and other treatment-related factors on mid-term to long-term cognitive sequelae in low-grade gliomas: a comparative study , in Lancet , vol. 360, n. 9343, 2 novembre 2002, pp. 1361-8, DOI : 10.1016/S0140-6736(02)11398-5 , PMID 12423981 .
- ^ a b ( EN ) Shaw E, Arusell R, Scheithauer B, O'Fallon J, O'Neill B, Dinapoli R, Nelson D, Earle J, Jones C, Cascino T, Nichols D, Ivnik R, Hellman R, Curran W, Abrams R (2002).
Prospective randomized trial of low- versus high-dose radiation therapy in adults with supratentorial low-grade glioma: initial report of a North Central Cancer Treatment Group/Radiation Therapy Oncology Group/Eastern Cooperative Oncology Group Study.
J Clin Oncol. 2002 May 1;20(9):2267-76. - ^ a b ( EN ) Karim AB, Maat B, Hatlevoll R, Menten J, Rutten EH, Thomas DG, Mascarenhas F, Horiot JC, Parvinen LM, van Reijn M, Jager JJ, Fabrini MG, van Alphen AM, Hamers HP, Gaspar L, Noordman E, Pierart M, van Glabbeke M (1996).
A randomized trial on doseresponse in radiation therapy of low-grade cerebral glioma: European Organization for Research and Treatment of Cancer (EORTC) Study 22844.
Int J Radiat Oncol Biol Phys. 1996 Oct 1;36(3):549-56. - ^ a b ( EN ) EG Shaw, B. Berkey, SW Coons, D. Brachman, JC Buckner, KJ Stelzer, GR Barger, PD Brown, MR Gilbert and M. Mehta (2006).
Initial report of Radiation Therapy Oncology Group (RTOG) 9802: prospective studies in adult lowgrade glioma (LGG). [ collegamento interrotto ]
J Clin Oncol. 2006;24(18s, pt I):63s. Abstract 1500. - ^ ( EN ) Pace A, Vidiri A, Galiè E, Carosi M, Telera S, Cianciulli AM, Canalini P, Giannarelli D, Jandolo B, Carapella CM (2003).
Temozolomide chemotherapy for progressive low-grade glioma: clinical benefits and radiological response.
Ann Oncol. 2003 Dec;14(12):1722-6. - ^ ( EN ) Neyns B, Sadones J, Chaskis C, De Ridder M, Keyaerts M, Veld PI, Michotte A (2005).
The role of chemotherapy in the treatment of low-grade glioma. A review of the literature.
Acta Neurol Belg. 2005 Sep;105(3):137-43. - ^ ( EN ) Pouratian N, Gasco J, Sherman JH, Shaffrey ME, Schiff D (2007).
Toxicity and efficacy of protracted low dose temozolomide for the treatment of low grade gliomas.
J Neurooncol. 2007 May;82(3):281-8. Epub 2006 Nov 3. - ^ ( EN ) Kaloshi G, Benouaich-Amiel A, Diakite F, Taillibert S, Lejeune J, Laigle-Donadey F, Renard MA, Iraqi W, Idbaih A, Paris S, Capelle L, Duffau H, Cornu P, Simon JM, Mokhtari K, Polivka M, Omuro A, Carpentier A, Sanson M, Delattre JY, Hoang-Xuan K (2007).
Temozolomide for low-grade gliomas: predictive impact of 1p/19q loss on response and outcome. Archiviato il 5 luglio 2008 in Internet Archive .
Neurology. 2007 May 22;68(21):1831-6. - ^ ( EN ) Liu R, Solheim K, Polley MY, Lamborn KR, Page M, Fedoroff A, Rabbitt J, Butowski N, Prados M, Chang SM (2008).
Quality of life in low-grade glioma patients receiving temozolomide. [ collegamento interrotto ] Neuro Oncol. 2009 Feb;11(1):59-68. Epub 2008 Aug 19. - ^ ( EN ) Buckner JC (2003).
Factors influencing survival in high-grade gliomas.
Semin Oncol. 2003 Dec;30(6 Suppl 19):10-4. - ^ ( EN ) Walker MD, Green SB, Byar DP, Alexander E Jr, Batzdorf U, Brooks WH, Hunt WE, MacCarty CS, Mahaley MS Jr, Mealey J Jr, Owens G, Ransohoff J 2nd, Robertson JT, Shapiro WR, Smith KR Jr, Wilson CB, Strike TA (1980).
Randomized comparisons of radiotherapy and nitrosoureas for the treatment of malignant glioma after surgery.
N Engl J Med. 1980 Dec 4;303(23):1323-9. - ^ ( EN ) Gregor A, Cull A (1996).
Radiotherapy for malignant glioma.
BMJ. 1996 Dec 14;313(7071):1500-1. - ^ ( EN ) Prados MD, Scott C, Curan WJ Jr, Nelson DF, Leibel S, Kramer S (1999).
Procarbazine, lomustine, and vincristine (PCV) chemotherapy for anaplastic astrocytoma: a retrospective review of radiation therapy oncology group protocols comparing survival with carmustine or PCV adjuvant chemotherapy.
J Clin Oncol. 1999 Nov;17(11):3389-95. - ^ ( EN ) Stewart LA (2002).
Chemotherapy in adult high-grade glioma: a systemic review and meta-analysis of individual patient data from 12 randomised trials.
Lancet. 2002 Mar 23;359(9311):1011-8. - ^ ( EN ) Medical Research Council Brain Tumor Working Party (2001).
Randomized trial of procarbazine, lomustine, and vincristine in the adjuvant treatment of high-grade astrocytoma: a Medical Research Council trial.
J Clin Oncol. 2001 Jan 15;19(2):509-18. - ^ ( EN ) Banna GL, Bettio D, Scorsetti M, Navarria P, Simonelli M, Rodriguez Baena R, Aimar E, Gaetani P, Colombo P, Rognone F, Santoro A (2006).
Administration of temozolomide during and after radiotherapy for newly diagnosed high-grade gliomas excluding glioblastoma multiforme.
J Neurooncol. 2007 Feb;81(3):323-5. Epub 2006 Sep 23. - ^ ( EN ) Yung WK, Prados MD, Yaya-Tur R, Rosenfeld SS, Brada M, Friedman HS, Albright R, Olson J, Chang SM, O'Neill AM, Friedman AH, Bruner J, Yue N, Dugan M, Zaknoen S, Levin VA (1999).
Multicenter phase II trial of temozolomide in patients with anaplastic astrocytoma or anaplastic oligoastrocytoma at first relapse.
J Clin Oncol. 1999 Sep;17(9):2762-71. Erratum in: J Clin Oncol 1999 Nov;17(11):3693. - ^ Martin V (2007).
FISH e tumori cerebrali. [ collegamento interrotto ]
ICP(Istituto Cantonale di Patologia) Locarno, 24 aprile 2007. Consultato il 12 marzo 2009. - ^ Traslocazione sbilanciata si ha quando a seguito di rottura e scambio di parti tra due cromosomi vi è perdita di materiale genetico e quindi danno genetico. (Cfr. http://www.laboratoriobiogen.it/lab/glossario.asp#T Archiviato il 29 settembre 2008 in Internet Archive ..) In questa operazione è coinvolto il centromero .
- ^ a b ( EN ) Jenkins RB, Blair H, Ballman KV, Giannini C, Arusell RM, Law M, Flynn H, Passe S, Felten S, Brown PD, Shaw EG, Buckner JC (2006).
A t(1;19)(q10;p10) mediates the combined deletions of 1p and 19q and predicts a better prognosis of patients with oligodendroglioma.
Cancer Res. 2006 Oct 15;66(20):9852-61. - ^ ( EN ) Hoang-Xuan K, Capelle L, Kujas M, Taillibert S, Duffau H, Lejeune J, Polivka M, Crinière E, Marie Y, Mokhtari K, Carpentier AF, Laigle F, Simon JM, Cornu P, Broët P, Sanson M, Delattre JY (2004).
Temozolomide as initial treatment for adults with low-grade oligodendrogliomas or oligoastrocytomas and correlation with chromosome 1p deletions.
J Clin Oncol. 2004 Aug 1;22(15):3133-8. - ^ ( EN ) Brada M, Viviers L, Abson C, Hines F, Britton J, Ashley S, Sardell S, Traish D, Gonsalves A, Wilkins P, Westbury C (2003).
Phase II study of primary temozolomide chemotherapy in patients with WHO grade II gliomas.
Ann Oncol. 2003 Dec;14(12):1715-21. - ^ ( EN ) Buckner JC, Gesme D Jr, O'Fallon JR, Hammack JE, Stafford S, Brown PD, Hawkins R, Scheithauer BW, Erickson BJ, Levitt R, Shaw EG, Jenkins R (2003).
Phase II Trial of Procarbazine, Lomustine, and Vincristine as Initial Therapy for Patients With Low-Grade Oligodendroglioma or Oligoastrocytoma: Efficacy and Associations With Chromosomal Abnormalities.
J Clin Oncol. 2003 Jan 15;21(2):251-5. - ^ ( EN ) Quinn JA, Reardon DA, Friedman AH, Rich JN, Sampson JH, Provenzale JM, McLendon RE, Gururangan S, Bigner DD, Herndon JE 2nd, Avgeropoulos N, Finlay J, Tourt-Uhlig S, Affronti ML, Evans B, Stafford-Fox V, Zaknoen S, Friedman HS (2003).
Phase II trial of temozolomide in patients with progressive low-grade glioma.
J Clin Oncol. 2003 Feb 15;21(4):646-51. - ^ ( EN ) Pace A, Vidiri A, Galiè E, Carosi M, Telera S, Cianciulli AM, Canalini P, Giannarelli D, Jandolo B, Carapella CM (2003).
Temozolomide chemotherapy for progressive low-grade glioma: clinical benefits and radiological response.
Ann Oncol. 2003 Dec;14(12):1722-6. - ^ ( EN ) Intergroup Radiation Therapy Oncology Group Trial 9402, Cairncross G, Berkey B, Shaw E, Jenkins R, Scheithauer B, Brachman D, Buckner J, Fink K, Souhami L, Laperierre N, Mehta M, Curran W (2006).
Phase III trial of chemotherapy plus radiotherapy compared with radiotherapy alone for pure and mixed anaplastic oligodendroglioma: Intergroup Radiation Therapy Oncology Group Trial 9402.
J Clin Oncol. 2006 Jun 20;24(18):2707-14. - ^ ( EN ) van den Bent MJ, Carpentier AF, Brandes AA, Sanson M, Taphoorn MJ, Bernsen HJ, Frenay M, Tijssen CC, Grisold W, Sipos L, Haaxma-Reiche H, Kros JM, van Kouwenhoven MC, Vecht CJ, Allgeier A, Lacombe D, Gorlia T (2006).
Adjuvant procarbazine, lomustine, and vincristine improves progression-free survival but not overall survival in newly diagnosed anaplastic oligodendrogliomas and oligoastrocytomas: a randomized European Organisation for Research and Treatment of Cancer phase III trial.
J Clin Oncol. 2006 Jun 20;24(18):2715-22. - ^ ( EN ) Cairncross G, Macdonald D, Ludwin S, Lee D, Cascino T, Buckner J, Fulton D, Dropcho E, Stewart D, Schold C Jr, et al (1994).
Chemotherapy for anaplastic oligodendroglioma. National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 1994 Oct;12(10):2013-21. - ^ Mielosoppressione . La diminuzione dell'attività delle cellule precursori del sangue, localizzate nel midollo osseo. I globuli rossi ei globuli bianchi del nostro sangue vengono generati dalle cellule staminali localizzate nel midollo osseo. Tali cellule generalmente hanno vita breve e devono essere rimpiazzate costantemente. Per fare ciò, le cellule staminali precursori si dividono molto rapidamente. Gli agenti chemioterapici, radioterapici e molti altri trattamenti anti-tumorali sono progettati per attaccare le cellule che si dividono rapidamente, e spesso inibiscono l'attività di tali cellule sane del midollo osseo. Molti effetti collaterali delle terapie anti-tumorali, come l'anemia e la diminuzione della capacità di combattere le infezioni (immunosoppressione) sono correlate agli effetti di tali trattamenti sulle cellule del midollo osseo.
Cancerquest Dictionary (2008).
Mielosoppressione. Archiviato il 14 luglio 2007 in Internet Archive .
Emory University. URL consultato il 20 marzo 2009. - ^ a b c ( EN ) Massimino M (2004).
Ependymoma
Orphanet Encyclopedia, February 2004.URL consultato il 21 marzo 2009. - ^ ( EN ) Centeno RS, Lee AA, Winter J, Barba D (1986).
Supratentorial ependymomas. Neuroimaging and clinicopathological correlation.
J Neurosurg. 1986 Feb;64(2):209-15. - ^ ( EN ) Gornet MK, Buckner JC, Marks RS, Scheithauer BW, Erickson BJ (1999).
Chemotherapy for advanced CNS ependymoma.
J Neurooncol. Jan 1999;45(1):61-67. - ^ ( EN ) Packer RJ, Cogen P, Vezina G, Rorke LB (1999).
Medulloblastoma: clinical and biologic aspects. Archiviato il 3 settembre 2009 in Internet Archive .
Neuro Oncol. 1999 Jul;1(3):232-50. - ^ ( EN ) Rohringer M, Sutherland GR, Louw DF, Sima AA (1989).
Incidence and clinicopathological features of meningioma.
J Neurosurg. 1989 Nov;71(5 Pt 1):665-72. - ^ ( EN ) Lieu AS, Howng SL (2000).
Intracranial meningiomas and epilepsy: incidence, prognosis and influencing factors.
Epilepsy Res. 2000 Jan;38(1):45-52. - ^ ( EN ) START Oncology in Europe (2009).
Meningioma. Archiviato il 6 luglio 2007 in Internet Archive .
URL consultato il 22 marzo 2009. - ^ ( EN ) Go RS, Taylor BV, Kimmel DW (1998).
The natural history of asymptomatic meningiomas in Olmsted County, Minnesota.
Neurology. 1998 Dec;51(6):1718-20. - ^ ( EN ) Pollock BE, Stafford SL, Utter A, Giannini C, Schreiner SA (2003).
Stereotactic radiosurgery provides equivalent tumor control to Simpson Grade 1 resection for patients with small- to medium-size meningiomas.
Int J Radiat Oncol Biol Phys. 2003 Mar 15;55(4):1000-5. - ^ ( EN ) Deckert M, Paulus W (2007). Malignant lymphomas , in
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds) (2007).
World Health Organization Classification of Tumours of the Central Nervous System.
IARC, Lyon ISBN 92-832-2430-2 . - ^ Janssen-Cilag SpA Divisione Tibotec
Conta CD4 & carica virale. Archiviato il 29 giugno 2009 in Internet Archive . URL consultato il 25 marzo 2009. - ^ a b c ( EN ) Batchelor T, Carson K, O'Neill A, Grossman SA, Alavi J, New P, Hochberg F, Priet R (2003).
Treatment of primary CNS lymphoma with methotrexate and deferred radiotherapy: a report of NABTT 96-07.
J Clin Oncol. 2003 Mar 15;21(6):1044-9. - ^ a b ( EN ) DeAngelis LM, Seiferheld W, Schold SC, Fisher B, Schultz CJ; Radiation Therapy Oncology Group Study 93-10 (2002).
Combination chemotherapy and radiotherapy for primary central nervous system lymphoma: Radiation Therapy Oncology Group Study 93-10.
J Clin Oncol. 2002 Dec 15;20(24):4643-8. - ^ a b ( EN ) Doolittle ND, Miner ME, Hall WA, Siegal T, Jerome E, Osztie E, McAllister LD, Bubalo JS, Kraemer DF, Fortin D, Nixon R, Muldoon LL, Neuwelt EA (2000).
Safety and efficacy of a multicenter study using intraarterial chemotherapy in conjunction with osmotic opening of the blood-brain barrier for the treatment of patients with malignant brain tumors.
Cancer. 2000 Feb 1;88(3):637-47. - ^ a b ( EN ) Patchell RA, Tibbs PA, Walsh JW, Dempsey RJ, Maruyama Y, Kryscio RJ, Markesbery WR, Macdonald JS, Young B (1990).
A randomized trial of surgery in the treatment of single metastases to the brain.
N Engl J Med. 1990 Feb 22;322(8):494-500. - ^ a b c ( EN ) Patchell RA, Tibbs PA, Regine WF, Dempsey RJ, Mohiuddin M, Kryscio RJ, Markesbery WR, Foon KA, Young B (1998).
Postoperative radiotherapy in the treatment of single metastases to the brain: a randomized trial.
JAMA. 1998 Nov 4;280(17):1485-9. - ^ ( EN ) Andrews DW, Scott CB, Sperduto PW, Flanders AE, Gaspar LE, Schell MC, Werner-Wasik M, Demas W, Ryu J, Bahary JP, Souhami L, Rotman M, Mehta MP, Curran WJ Jr (2004).
Whole brain radiation therapy with or without stereotactic radiosurgery boost for patients with one to three brain metastases: phase III results of the RTOG 9508 randomised trial.
Lancet. 2004 May 22;363(9422):1665-72. - ^ ( EN ) Kondziolka D, Patel A, Lunsford LD, Kassam A, Flickinger JC (1999).
Stereotactic radiosurgery plus whole brain radiotherapy versus radiotherapy alone for patients with multiple brain metastases.
Int J Radiat Oncol Biol Phys. 1999 Sep 1;45(2):427-34. - ^ ( EN ) Muacevic A, Wowra B, Kreth FW, Tonn JC (2006).
A randomized trial of surgery and radiotherapy versus radiosurgery alone in the treatment of single metastasis to the brain [abstract].
German Med Sci. 2006. Abstract OP256. - ^ ( EN ) Bindal AK, Bindal RK, Hess KR, Shiu A, Hassenbusch SJ, Shi WM, Sawaya R (1996).
Surgery versus radiosurgery in the treatment of brain metastasis. [ collegamento interrotto ]
J Neurosurg. 1996 May;84(5):748-54. - ^ ( EN ) O'Neill BP, Iturria NG, Link MJ, Pollock BE, Ballman KV, O'Fallon JR (2003).
A comparison of surgical resection and stereotactic radiosurgery in the treatment of solitary brain metastases.
Int J Radiat Oncol Biol Phys. 2003 Apr 1;55(5):1169-76. - ^ ( EN ) Pollock BE, Brown PD, Foote RL, Stafford SL, Schomberg PJ (2003).
Properly selected patients with multiple brain metastases may benefit from aggressive treatment of their intracranial disease.
J Neurooncol. 2003 Jan;61(1):73-80. - ^ ( EN ) Antonadou D, Paraskevaidis M, Sarris G, Coliarakis N, Economou I, Karageorgis P, Throuvalas N (2002).
Phase II randomized trial of temozolomide and concurrent radiotherapy in patients with brain metastases.
J Clin Oncol. 2002 Sep 1;20(17):3644-50. - ^ ( EN ) Mehta MP, Rodrigus P, Terhaard CH, Rao A, Suh J, Roa W, Souhami L, Bezjak A, Leibenhaut M, Komaki R, Schultz C, Timmerman R, Curran W, Smith J, Phan SC, Miller RA, Renschler MF (2003).
Survival and neurologic outcomes in a randomized trial of motexafin gadolinium and whole-brain radiation therapy in brain metastases.
J Clin Oncol. 2003 Jul 1;21(13):2529-36 - ^ ( EN ) Suh JH, Stea B, Nabid A, Kresl JJ, Fortin A, Mercier JP, Senzer N, Chang EL, Boyd AP, Cagnoni PJ, Shaw E (2006).
Phase III study of efaproxiral as an adjunct to whole-brain radiation therapy for brain metastases.
J Clin Oncol. 2006 Jan 1;24(1):106-114. Epub 2005 Nov 28. - ^ ( EN ) DeAngelis LM, Loeffler JS, Adam N. Mamelak AN (2007).
Primary and Metastatic Brain Tumors .
In Pazdur R, Coia LR, Hoskins WJ, and Wagman LD (2007). Cancer Management: A Multidisciplinary Approach, 10th Edition .
URL consultato il 30-03-2009. - ^ Erbetta A, Lauria G, Sghirlanzoni A (2006).
Complicazioni della chemioterapia.
in: Caraceni A, Sghirlanzoni A, Simonetti F (2006). Le complicazioni neurologiche in oncologia.
Springer 2006 ISBN 978-88-470-0439-9 .
Bibliografia
- ( EN ) Lisa M. DeAngelis (2001). Brain Tumors . , N Engl J Med. 2001 Jan 11;344(2):114-23.
- ( EN ) Primary and metastatic brain tumors (2010). Primary and metastatic brain tumors . In Pazdur R, Wagman LD, Camphausen KA, Hoskins WJ (2009). Cancer Management: A Multidisciplinary Approach, 12th Edition. CancerNetwork. Updated: 2010 Mar 11.
- ( EN ) Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds) (2007). World Health Organization Classification of Tumours of the Central Nervous System . IARC, Lyon ISBN 92-832-2430-2 .
- ( EN ) Packer RJ (2008). Childhood brain tumors: accomplishments and ongoing challenges [ collegamento interrotto ] . J Child Neurol. 2008 Oct;23(10):1122-7.
- ( EN ) Wen PY, Kesari S (2008). Malignant gliomas in adults . N Engl J Med. 2008 Jul 31;359(5):492-507. Erratum in: N Engl J Med. 2008 Aug 21;359(8):877.
Anatomia patologica e medicina interna
- Robbins e Cotran, Le basi patologiche delle malattie , 7ª ed., Torino-Milano, Elsevier Masson, 2008, ISBN 978-88-85675-53-7 .
- Mariuzzi, Anatomia patologica e correlazioni anatomo-cliniche , Padova, Piccin, 2006, ISBN 978-88-299-1769-3 .
- Harrison, Principi di Medicina Interna , 16ª ed., New York-Milano, McGraw-Hill, 2006, ISBN 88-386-2459-3 .
Farmacologia
- Brunton, Lazo, Parker, Goodman & Gilman - Le basi farmacologiche della terapia , 11ª ed., McGraw Hill, 2006, ISBN 978-88-386-3911-1 .
- Bertram G. Katzung, Farmacologia generale e clinica , Padova, Piccin, 2006, ISBN 88-299-1804-0 .
Neurologia
- C. Loeb, E. Favale, Neurologia di Fazio Loeb , Roma, Società Editrice Universo, 2003, ISBN 88-87753-73-3 .
- B. Bergamasco, R. Mutani, La neurologia di Bergamini , Torino, Cortina, 2007, ISBN 88-8239-120-5 .
- Allan H. Ropper, Robert H. Brown, Adams & Victor - Principi di neurologia , Milano - New York, McGraw-Hill Companies, 2006, ISBN 88-386-3909-4 .
Linee guida
- ( EN ) US Department of Health & Human Services. National Guideline Clearinghouse (2009). [ https://www.guideline.gov/search/search.aspx?term=brain+tumor¨s=1 [ collegamento interrotto ] Brain tumor] . URL consultato il 21 gennaio 2011.
- ( EN ) National Institute for Health and Clinical Excellence (2006). Service guidance for improving outcomes for people with brain and other central nervous system tumours . URL consultato il 21 gennaio 2011.
- ( EN ) American Academy of Family Physicians (2008). Primary Brain Tumors in Adults . A cura di Chandana SR, Movva S, Arora M, Singh T. URL consultato il 21 gennaio 2011.
- ( EN ) National Cancer Institute (2010). General Information About Adult Brain Tumors . URL consultato il 21 gennaio 2011.
Linee guida italiane
- AIOM - Associazione Italiana di Oncologia Medica (2009). Neoplasie cerebrali . A cura di Brandes AA, Calbucci F, Leonardi M, Reni M, Spagnolli F, Tosoni A, Labianca R, Ferrarese F, Carapella C. URL consultato il 21 gennaio 2011.
- Ministero della Salute. Basi Scientifiche Linee Guida (2007). Tumori cerebrali . A cura di Rosella Silvestrini. URL consultato il 21 gennaio 2011.
Voci correlate
- Classificazione dei tumori del sistema nervoso centrale
- Gradazione dei tumori del sistema nervoso centrale
- Sistema nervoso
- Tumore
Collegamenti esterni
Vengono qui riportati i link alle maggiori riviste mediche specializzate.
- Acta Neuropathologica , su springerlink.com . URL consultato il 16 gennaio 2011 (archiviato dall' url originale il 6 dicembre 2009) .
- American Journal of Neuroradiology , su ajnr.org .
- Annals of Neurology [ collegamento interrotto ] , su www3.interscience.wiley.com .
- Archives of Neurology , su archneur.ama-assn.org .
- Brain , su brain.oxfordjournals.org .
- Brain Pathology [ collegamento interrotto ] , su www3.interscience.wiley.com .
- Brain Tumor Pathology [ collegamento interrotto ] , su springerlink.com .
- British Journal of Neurosurgery , su informahealthcare.com .
- Child's Nervous System , su springerlink.com . URL consultato il 16 gennaio 2011 (archiviato dall' url originale il 3 ottobre 2012) .
- Clinical Neuropathology , su dustri.com .
- Glia [ collegamento interrotto ] , su www3.interscience.wiley.com .
- International Journal of Radiation Oncology, Biology, Physics , su sciencedirect.com .
- Journal of Neurology [ collegamento interrotto ] , su springerlink.com .
- Journal of Neurology, Neurosurgery, and Psychiatry , su jnnp.bmj.com .
- Journal of Neuro-Oncology , su springerlink.metapress.com .
- Journal of Neuropathology and Experimental Neurology , su journals.lww.com .
- Journal of Neurosurgery , su thejns.org .
- The Lancet Neurology , su thelancet.com .
- Nature Reviews Neurology , su nature.com .
- Neurological Research , su ingentaconnect.com . URL consultato il 16 gennaio 2011 (archiviato dall' url originale l'11 febbraio 2011) .
- Neurology , su neurology.org .
- Neuroradiology [ collegamento interrotto ] , su springerlink.com .
- Neuro-Oncology , su neuro-oncology.oxfordjournals.org .
- Neurosurgery , su journals.lww.com .
- Neurosurgical Focus , su thejns.org .
- Pediatric Neurology , su journals.elsevierhealth.com .
- Pediatric Neurosurgery , su content.karger.com .
- Revista de neurologia , su revneurol.com .
- Surgical Neurology , su journals.elsevierhealth.com . URL consultato il 16 gennaio 2011 (archiviato dall' url originale il 23 luglio 2007) .
Gli articoli di rilevanza in materia pubblicati altrove ( Cancer , JAMA , JCO , NEJM , ecc.) sono normalmente segnalati dalla newsletter elettronica
- Current Neuro-Oncology , su brainlife.org . URL consultato il 27 giugno 2011 (archiviato dall' url originale il 25 luglio 2011) .