boala Creutzfeldt-Jakob
| boala Creutzfeldt-Jakob | |
|---|---|
 | |
| Specialitate | neurologie |
| Etiologie | prion |
| Clasificare și resurse externe (EN) | |
| OMIM | 123400 și 123400 |
| Plasă | D007562 |
| MedlinePlus | 000788 |
| eMedicină | 1169688 |
| Eponime | |
| Hans Gerhard Creutzfeldt Alfons Maria Jakob | |
Boala Creutzfeldt-Jakob (CJD), descrisă inițial în anii '20 ai secolului XX de Hans Gerhard Creutzfeldt și Alfons Maria Jakob , este o boală neurodegenerativă rară, care duce la o formă de demență fatală progresivă. [1]
Sindromul clinic se caracterizează prin deficite polisectoriale predominant corticale cu pierderi de memorie , modificări ale personalității , halucinații , disartrie , mioclonie , rigiditate posturală și convulsii . Boala Creutzfeldt-Jakob este cea mai frecventă formă de encefalopatie spongiformă umană. [2] La nivel histologic există formarea de microvacuulații ale țesutului cerebral care își asumă o structură spongioasă, datorită pierderii progresive a neuronilor cauzată de alterarea unei proteine de membrană, exprimată în principal în celulele sistemului nervos și a reticulul- endotelial , prionul . Boala Creutzfeldt-Jakob aparține grupului de encefalopatii spongiforme transmisibile , un tip de boală neurodegenerativă datorată prezenței prionilor . [3] Prionul în forma sa modificată (PrP Sc ) demonstrează capacitatea infecțioasă, adică se poate răspândi acționând asupra formei native (PrP c ), făcând ca boala Creutzfeldt-Jakob să fie inclusă în encefalopatiile transmisibile, chiar dacă este nu este considerat contagios în sensul tradițional. [4]
Incidența CJD a rămas relativ constantă în ultimii 80 de ani, de ordinul 1-2 / 1.000.000 / an. El primește titluri după ce a descris primele cazuri ale unei forme variante , chiar mai rare, legate de epidemia de encefalopatie spongiformă bovină , așa-numita boală „vaca nebună”. [5] [6]
Istorie
Boala a fost descrisă pentru prima dată în 1920 de neurologul german Hans Gerhard Creutzfeldt și la scurt timp de Alfons Maria Jakob , de unde și numele Creutzfeldt-Jakob. Unele dintre descoperirile clinice descrise în lucrările lor timpurii nu corespund criteriilor actuale pentru boală și s-a emis ipoteza că cel puțin doi dintre pacienții observați în studiile inițiale sufereau de fapt de o boală diferită. O primă descriere a formei familiale este dată de neurologul și psihiatrul german Friedrich Meggendorfer (1880-1953). [7] [8]
Epidemiologie

Deși CJD a fost cea mai frecventă boală umană cauzată de prioni, prezentarea sa este încă rară, apărând la aproximativ un milion de oameni în fiecare an. De obicei, afectează persoanele cu vârste cuprinse între 45 și 75 de ani și apare cel mai frecvent la persoanele cu vârste cuprinse între 60 și 65 de ani. Excepția de la aceasta este varianta mai nouă (vMCJ), care apare la persoanele mai tinere.
Centrele Statelor Unite pentru Controlul și Prevenirea Bolilor (CDC) monitorizează prezența bolii în țară, prin analize periodice ale datelor naționale privind mortalitatea . Conform CDC:
- CJD s-a produs la nivel mondial la o rată de aproximativ 1 pe milion de populație pe an.
- Pe baza supravegherii mortalității înregistrate între 1979 și 1994, incidența anuală a CJD a rămas stabilă la aproximativ 1 caz pe milion de persoane în Statele Unite.
- În Statele Unite, decesele prin CJD în rândul persoanelor cu vârsta sub 30 de ani au avut loc într-un procent foarte scăzut.
- Boala a fost frecvent întâlnită la pacienții cu vârsta cuprinsă între 55 și 65 de ani, dar, în unele cazuri, ar putea fi la persoanele cu vârsta peste 90 de ani și cu vârsta sub 55 de ani.
- În mai mult de 85% din cazuri, durata bolii, după apariția simptomelor, a fost mai mică de un an ( mediană : patru luni), ducând la moarte sigură. [1] [9]
În ceea ce privește noua variantă , din 1996 până în martie 2014 numărul total de cazuri s-a soluționat la 225, dintre care 177 numai în Marea Britanie. [10] Graficul din dreapta descrie națiunile afectate de epidemia de formă variantă și omologul său bovin.
Clasificare
Tipurile de CJD includ:
- Formă sporadică (SMCJ) [11]
- Forma familială (fMCJ) [12]
- Forma iatrogenă (iMCJ) [13]
- Varianta nouă (vMCJ) [14] Identificată pentru prima dată în 1996. [15]
semne si simptome
Primul simptom al CJD este o demență rapidă și progresivă care duce la pierderea memoriei, modificări ale personalității și halucinații . Acest lucru este însoțit de probleme fizice, cum ar fi tulburări de vorbire, mișcări rapide involuntare ( mioclonus ), disfuncții de echilibru și coordonare ( ataxie ), modificări ale mersului, postură rigidă și convulsii . Durata bolii variază foarte mult, ducând la deces în câteva luni sau câteva săptămâni (Johnson, 1998). La unele persoane, simptomele pot continua ani de zile. La majoritatea pacienților, aceste simptome sunt urmate de mișcări involuntare și de apariția unui EEG atipic. Majoritatea pacienților mor în decurs de șase luni de la debut, adesea din cauza infecțiilor intercurente, cum ar fi pneumonia, din cauza reflexului de tuse deteriorat. Aproximativ 15% dintre pacienți supraviețuiesc timp de doi sau mai mulți ani. [16]
Simptomele CJD sunt cauzate de moartea progresivă a celulelor nervoase din creier, care este asociată cu formarea plăcilor amiloide datorate prionilor. Când țesutul cerebral al unui pacient cu boala Creutzfeldt-Jakob este examinat la microscop , pot fi văzute multe găuri mici, unde au murit zone întregi ale celulelor nervoase. Cuvântul „spongiform” din „encefalopatii spongiforme” se referă la aspectul spongios al țesutului cerebral.
Etiopatogenie
Encefalopatiile spongiforme transmisibile sunt boli cauzate de prioni. Bolile sunt uneori numite „boli prionice”. Alte boli prionice care afectează oamenii includ boala Gerstmann-Sträussler-Scheinker (GSS), insomnia familială fatală (IFF) și kuru , precum și encefalopatia spongiformă bovină (ESB, cunoscută sub denumirea de vacă nebună) la bovine , boala de pierdere cronică (CWD), și scrapie la oi. Boala Alpers a nou-născuților este, de asemenea, considerată a fi o encefalopatie spongiformă transmisibilă cauzată de un prion. [17] [18]
Prionul despre care se crede că este cauza Creutzfeldt-Jakob are cel puțin două conformații stabile. Primul, în starea sa nativă, este solubil în apă și este prezent în celulele sănătoase. Începând cu 2007, se crede că funcția sa biologică este implicată probabil în transportul sau semnalizarea transmembranară. Cealaltă stare conformațională este relativ insolubilă în apă și formează ușor agregate proteice.
De asemenea, oamenii pot dobândi boala genetic printr-o mutație a genei care codifică proteina prionică (PRNP). Cu toate acestea, acest lucru apare doar în 5-10% din toate cazurile de boală Creutzfeldt-Jakob.
Prionul Creutzfeldt-Jakob este periculos deoarece, în timpul bolii, promovează plierea incorectă a proteinelor. [19] Numărul de molecule de proteine pliate greșit crește exponențial și procesul formează o cantitate mare de proteine insolubile în celulele afectate. Această masă de proteine pliate greșit distruge funcția celulară și provoacă moartea celulelor. Odată ce prionul este transmis, proteinele defecte invadează creierul.
Stanley B. Prusiner de la Universitatea din California , San Francisco (UCSF) a primit Premiul Nobel pentru fiziologie și medicină din 1997 pentru descoperirea prionilor. De mai bine de un deceniu, Laura Manuelidis, neuropatologă a Universității Yale , s-a angajat în explicația dificilă a bolii. În ianuarie 2007, ea și colegii ei au publicat un articol susținând că au găsit particule asemănătoare virusului (dar nu au găsit acizi nucleici în ele) în mai puțin de 10% din celulele de șoarece infectate cu agentul uman al bolii Creutzfeldt-Jakob. [20]
Transmisie
Proteina defectă poate fi transmisă de substanțe contaminate colectate în creierul uman: imunoglobuline , grefe corneene , grefe durale sau prin implanturi de electrod (forma dobândită sau iatrogenă: iMCJ), dar poate fi moștenită și (formă ereditară sau familială: fMCJ), sau poate apărea pentru prima dată la pacient (formă sporadică: MCJ). În forma ereditară, apare o mutație în gena pentru PrP, PRNP . 10-15% din cazurile de BCJ sunt ereditare.
Boala a fost, de asemenea, indicată ca urmare a utilizării hormonului de creștere uman obținut din glandele pituitare ale persoanelor care au murit din cauza bolii Creutzfeldt-Jakob [21], deși incidența cunoscută pentru această cauză este mai degrabă (începând din aprilie 2004) mic. În Statele Unite, medicamentul care folosește hormonul cadaveric a fost retras în 1985 , punând capăt acestui tip de transmitere în țară.
Se crede că oamenii pot contracta boala consumând carne de la animale infectate cu forma bovină a bolii. [18] [22]
Canibalismul a fost, de asemenea, implicat ca o cauză de transmitere a prionilor anormali, provocând o boală cunoscută sub numele de kuru , întâlnită mai ales în rândul femeilor și copiilor din populația Fore din Papua Noua Guinee . În timp ce bărbații din trib mănâncă corpul decedatului și rareori contractă boala, femeile și copiii, care au mâncat părți mai puțin dorite ale corpului, inclusiv creierul, au fost de 8 ori mai multe șanse decât bărbații să contracteze kuru din țesutul infectat.
Prionii, agentul infecțios al BCJ, nu pot fi inactivați prin proceduri de sterilizare post-chirurgicală. Organizația Mondială a Sănătății și Centrele pentru Controlul și Prevenirea Bolilor recomandă ca instrumentele utilizate în astfel de cazuri să fie distruse imediat după utilizare. Cu toate acestea, nu s-au raportat cazuri de transmitere iatrogenă a CJD de la adoptarea procedurilor actuale de sterilizare și în niciun caz niciunul din 1979. [23] [24] [25]
Restricții pentru donatorii de sânge
În 2004, un nou raport publicat în revista medicală The Lancet a arătat că vMCJ poate fi transmis prin transfuzii de sânge . [26] Descoperirea i-a alarmat pe oficialii din domeniul sănătății, deoarece acest lucru ar putea duce la o epidemie mare în viitor. Nu există niciun test pentru a determina dacă un donator de sânge este infectat cu vMCJ. Ca reacție la acest raport, guvernul britanic a împiedicat pe oricine care a primit o transfuzie de sânge, începând din ianuarie 1980, să doneze sânge. [27] Începând din 1999, în Marea Britanie a fost interzisă utilizarea sângelui din Marea Britanie pentru a produce produse fracționare, cum ar fi albumina . [28]
La 28 mai 2002 , Food and Drug Administration a instituit o regulă care exclude din donație pe oricine a petrecut cel puțin șase luni în anumite țări europene (sau trei luni în Marea Britanie), din 1980 până în 1996. Având în vedere numărul mare de militari americani și AI membrii familiei lor care locuiesc în Europa, era de așteptat ca peste 7% din donatori să fie opriți din cauza politicii. Modificările ulterioare aduse acestei politici au facilitat restricția pentru un total cumulativ de cinci ani sau mai mult de la călătoriile civile în țările europene (șase luni sau mai mult, dacă sunt militare). Cu toate acestea, restricția de călătorie de trei luni în Regatul Unit nu s-a schimbat. [29]
În Singapore, Crucea Roșie exclude potențialii donatori care au petrecut o perioadă combinată de trei luni sau mai mult în Marea Britanie între 1980 și 1996.
În Noua Zeelandă , oricui a locuit în Marea Britanie, Franța sau Republica Irlanda pentru un total de șase luni sau mai mult între ianuarie 1980 și decembrie 1996 i s-a interzis definitiv donarea de sânge. Oricine a primit o transfuzie de sânge în aceste țări din ianuarie 1980 este, de asemenea, interzis definitiv.
Reglementări similare sunt de asemenea în vigoare în Germania și Italia, unde oricui a petrecut șase luni sau mai mult din viață în Marea Britanie între ianuarie 1980 și decembrie 1996 i se interzice în permanență să doneze sânge. [30]
În Polonia , oricine a petrecut șase sau mai multe luni cumulativ între 1 ianuarie 1980 și 31 decembrie 1996 în Regatul Unit, Irlanda sau Franța este exclus definitiv din donație. [31]
În Elveția, posibilitatea donării de sânge este permanent exclusă pentru cei care au primit o transfuzie de sânge integral de la 1 ianuarie 1980 și au locuit în Regatul Unit și, prin urmare, Anglia, Țara Galilor, Scoția, Irlanda de Nord, Insula Man, Insulele Canalului Mânecii, Insulele Falkland și Gibraltar pentru mai mult de șase luni între 1980 și 1996. [32]
Diagnostic

Un diagnostic de BCJ este suspectat atunci când se găsesc simptome și semne clinice tipice, cum ar fi demența rapid progresivă cu mioclonus . Analize suplimentare pot susține această suspiciune de diagnostic, inclusiv:
- Electroencefalografia , uneori prezintă vârfuri trifazice caracteristice
- Analiza lichidului cefalorahidian pentru detectarea proteinei 14-3-3 și pentru determinarea nivelurilor de tau total și fosforilat
- Rezonanța magnetică a creierului , arată adesea intensitate ridicată a semnalului în nucleul caudat și putamen bilateral, sau la nivel cortical în imaginile ponderate DWI ( D iffusion- W opted I maging) sau cu metoda FLAIR ( Fl uid A ttenuated I nversion R ecparmi).
- Un studiu din 2010 și 2011 a identificat un posibil test de sânge pentru BCJ. Testul încearcă să identifice prionul responsabil de boală. [33]
Metoda DWI oferă cele mai sensibile imagini pentru suspiciunea de BCJ, aproximativ 80% dintre pacienți îndeplinind criteriile radiologice. [34] În aproximativ 24% din cazuri, hiperintensitatea este identificabilă doar la nivel cortical (așa-numitul semn „panglică corticală”, datorită implicării elective a substanței gri), în 68% anomalii ale nucleilor corticali și bazali și în 5%, anomalii numai ale nucleilor bazei. [35] Implicarea talamusului poate fi găsită în CJD și în special în vMCJ. [36]
Tabloul clinic pentru CJD este variat și nu permite un diagnostic univoc, care necesită, de asemenea, criterii instrumentale sau de LCR. Cele mai recente criterii elaborate de criteriile consorțiului RMN-CJD pentru boala sporadică Creutzfeldt - Jakob includ următoarele
- Semne clinice
- Demenţă
- Semne cerebeloase sau vizuale
- Semne piramidale sau extrapiramidale
- Mutism ackinetic
- Examinări instrumentale
MCJ Probabil: două elemente de 1. și cel puțin unul din 2
CJD Posibil: două elemente de la 1. și durata bolii de la debut până la deces <2 ani [34]
În ultimii ani, diferite studii au arătat că markerul tumoral specific neuronului ( Enolaza 2 ) este adesea crescut în cazurile de BCJ, totuși utilitatea sa de diagnostic este văzută mai ales atunci când este combinată cu un test de proteine 14-3-3. [37] Începând cu 2012, testele de screening pentru identificarea persoanelor infectate asimptomatic, care urmează să fie utilizate de exemplu pentru donatorii de sânge , nu sunt încă disponibile, deși au fost propuse și evaluate diferite metode. [38]

În 2010, un grup de cercetători din New York a identificat PrP Sc, deși inițial prezent într-o singură parte din 100 miliarde (10 −11 ) de țesut cerebral . Metoda combină amplificarea cu o nouă tehnologie numită Surround Optical Fiber Immunoassay (SOFIA) și câțiva anticorpi specifici împotriva PrP Sc . După amplificare, PrP Scs sunt concentrate, probele sunt apoi marcate cu un colorant fluorescent folosind un anticorp specific și în cele din urmă încărcate într-un micro-capilar. Acest tub este plasat într-un aparat construit special, astfel încât să fie complet înconjurat de fibre optice pentru a capta toată lumina emisă atunci când colorantul este excitat cu un laser .
Cercetătorii și-au testat metoda pe probe de sânge de oaie aparent sănătoase, care au continuat să dezvolte scrapie . Creierul animalelor a fost analizat atunci când toate simptomele au devenit evidente. Rezultatele au arătat în mod clar că PrP Sc ar putea fi detectat în sângele animalelor înainte de apariția simptomelor. După dezvoltarea și testarea ulterioară a acestei metode, aceasta poate deveni de mare folos ca test de screening pentru CJD. [39] [40]
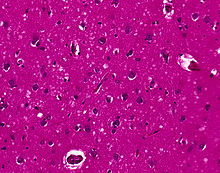
Diagnosticul vMCJ poate fi susținut de o biopsie a amigdalelor dacă se suspectează vMCJ, deoarece poate conține o cantitate semnificativă de PrP Sc . Biopsia țesutului cerebral este testul de diagnostic definitiv în toate celelalte forme de CJD și în cazul biopsiei amigdaliene negative și în vMCJ. Datorită invazivității sale, biopsia nu se efectuează dacă pe baza constatărilor anterioare suspiciunea clinică este foarte mare sau prea mică. O biopsie negativă nu exclude CJD, deoarece poate fi predominantă într-o anumită parte a creierului. [41]
Materia cenușie cerebrală își asumă aspectul histologic spongiform clasic: prezența a numeroase vacuole rotunde, cu aspect vitros sau eozinofil , cu tendință de fuziune și cu dimensiuni variabile între 1 și 50 micrometri, situate în toate cele șase straturi corticale din cortexul cerebral sau cu afectarea stratului cerebelos. De asemenea, pot fi observate pierderea neuronală și glioza . [42] Plăci de substanță amiloidă pot fi găsite în neocortex în cazurile unei noi variante a CJD.
Din păcate, vacuolarea este prezentă și în alte stări patologice. Vacuolarea corticală pe scară largă se observă și în boala Alzheimer, iar vacuolația corticală superficială apare în ischemii și demență frontotemporală. Aceste vacuole par clare. Vacuolele mai mari, care înconjoară neuronii, vasele și glia , sunt un posibil artefact de procesare. [41]
- Caracteristici clinice și patologice: [43]
| Caracteristică | MCJ clasic | Varianta MCJ |
|---|---|---|
| Vârsta medie la deces | 68 de ani | 28 de ani |
| Durata medie a bolii | 4–5 luni | 13-14 luni |
| Semne și simptome clinice | Demenţă; semne neurologice timpurii | Simptome psihiatrice / comportamentale; disestezie dureroasă; semne neurologice întârziate |
| Unde ascuțite periodic pe electroencefalogramă | Uneori prezent | Adesea absent |
| Hiperintensitatea semnalului în nucleul caudat și putamen sau la nivelul benzii corticale în secvențe DWI și / sau FLAIR | Adesea prezent | Adesea prezent |
| Analiza imunohistochimică a țesutului cerebral | Acumulări variabile | Acumularea marcată de proteine prionice rezistente la protează |
| Prezența agentului în țesutul ganglionar | Nu este ușor de detectat | Detectat cu ușurință |
| Prezența plăcilor amiloide în țesutul nervos | Ele pot fi prezente | Ele pot fi prezente |
- Un semnal anormal în talamusul posterior în imagistica prin rezonanță magnetică a creierului (IRM) ponderat T2, DWI și FLAIR , într-un cadru clinic adecvat, este foarte specific pentru vMCJ. (Sursa: CDC)
Tratament
Nu s-a demonstrat niciun tratament eficient pentru BCJ, boala este întotdeauna fatală și căutarea unui remediu continuă. Un tratament experimental a fost încercat unui adolescent din Irlanda de Nord , Jonathan Simms, în ianuarie 2003. [44] Medicamentul, numit pentosan polisulfester (PPS), este utilizat în general pentru tratarea cistitei interstițiale și este infuzat în ventriculul lateral al creierului pacientului. Medicamentul nu pare să poată opri boala și funcția creierului și a țesuturilor continuă să fie afectată. Cu toate acestea, se crede că tratamentul încetinește progresia bolii altfel incurabile și ar fi putut contribui la supraviețuirea mai lungă decât se aștepta a celor șapte pacienți studiați.[45] Experții Departamentului Sănătății din Regatul Unit nu cred suficiente date pentru a evalua utilizarea polifosfatului pentosan și recomandă cercetări suplimentare pe animale de laborator. [46] În 2007, analiza tratamentului a 26 de pacienți cu SPP nu a găsit dovezi de eficacitate. [47]
Oamenii de știință au fost pionieri în utilizarea interferenței ARN pentru a încetini progresia scrapiei la șoareci. Este puțin probabil ca această cercetare să conducă la terapia umană timp de mulți ani. [48]
Atât amfotericina B, cât și doxorubicina au fost studiate ca potențial eficiente împotriva CJD, dar nu există încă dovezi puternice că oricare dintre medicamente este eficient în oprirea bolii. Un alt studiu a fost încercat cu alte medicamente, dar niciunul nu a fost eficient. Cu toate acestea, există medicamente pentru a reduce suferința și includ valproat , un anticonvulsivant și clonazepam , pentru a reduce spasmele musculare [16]
Prognoză
Deoarece nu există o vindecare definitivă, prognosticul este slab în toate cazurile. 85% mor în doi ani, restul de 15% supraviețuiesc mai mult.
Aspecte speciale
În Italia este bine cunoscut embargoul asupra produselor britanice din carne de vită, care a durat aproximativ 10 ani, din 1996 până în 2006 , în urma epidemiei severe de CJD bovină din Marea Britanie în 1995 . Pentru a evita posibilitatea transmiterii CJD, directivele Ministerului Sănătății s-au mutat și în domeniul medicinei transfuzionale, reiterând în repetate rânduri obligația de a exclude de la donația de plasmă și măduvă osoasă pe cei care au primit implanturi de cornee sau care au avut cazuri de SJC sau insomnie fatală în familie în familia lor .
Notă
- ^ a b CJD (Boala Creutzfeldt-Jakob, clasică) , pe cdc.gov , Centers for Disease Control and Prevention, 26 februarie 2008. Accesat la 20 iunie 2009 (arhivat din original la 6 mai 2009) .
- ^ Boala Creutzfeldt-Jakob , pe Orphanet .
- ^ Creutzfeldt-Jakobs sjukdom , în Socialstyrelsen - Suedia . Adus pe 2 octombrie 2017 (arhivat din original la 20 septembrie 2017) .
- ^ Boala Creutzfeldt Jakob , despre Organizația Națională a Bolilor Rare - SUA .
- ^ VeriMed Healthcare Network, revizuit de: David C. Dugdale, Luc Jasmin, David Zieve, Boala Creutzfeldt-Jakob: Encefalopatie spongiformă transmisibilă; vCJD; CJD; Boala Jacob-Creutzfeldt , Biblioteca Națională de Medicină a SUA, 26 septembrie 2011. Accesat la 25 aprilie 2012 .
- ^ Paul Brown, Encefalopatia spongiformă bovină și varianta bolii Creutzfeldt-Jakob , BMJ , 4 iulie 2001. Accesat la 23 februarie 2011 .
- ^ Meggendorfer F. Klinische und genealogische Beobachtungen bei einem Fall von spastischer Pseudokosklerose Jakobs. Z Neurol Psychiatry 1930; 128: 337–41
- ^ Gambetti P, Kong Q, Zou W, Parchi P, Chen SG, CJD sporadică și familială: clasificare și caracterizare , în Br. Med. Bull. , vol. 66, 2003, pp. 213–39, DOI : 10.1093 / bmb / 66.1.213 , PMID 14522861 .
- ^ vCJD (Variant Creutzfeldt-Jakob Disease) , pe cdc.gov , Centers for Disease Control and Prevention, 4 ianuarie 2007. Accesat la 20 iunie 2009 (arhivat din original la 7 mai 2009) .
- ^ Unitatea Națională de Supraveghere a Bolilor Creutzfeldt-Jakob (NCJDSU) Total cazuri vCJD după țară Arhivat 26 februarie 2015 în Internet Archive ., Martie 2014
- ^ Niimi Y, Iwasaki Y, Umemura T, boala Creutzfeldt-Jakob sporadică de tip MM2-corticală cu patologie corticală cerebrală în stadiu incipient care prezintă un curs clinic rapid progresiv [ link rupt ] , în Neuropatologie , vol. 28, nr. 6, decembrie 2008, pp. 645–51, DOI : 10.1111 / j.1440-1789.2008.00904.x , PMID 18410280 .
- ^ Wang XF, Dong CF, Zhang J, proteinele tau umane formează complexe cu PrP și unele mutante PrP legate de GSS și fCJD posedă activități de legare mai puternice cu tau in vitro , în Mol. Cell. Biochem. , vol. 310, 1-2, martie 2008, pp. 49–55, DOI : 10.1007 / s11010-007-9664-6 , PMID 18038270 .
- ^ Hamaguchi T, Noguchi-Shinohara M, Nozaki I, Riscul bolii iatrogene Creutzfeldt-Jakob prin proceduri medicale și chirurgicale , în Neuropatologie , vol. 29, nr. 5, octombrie 2009, pp. 625-31, DOI : 10.1111 / j.1440-1789.2009.01023.x , PMID 19659942 .
- ^ Jones M, Peden AH, CV Prowse, Amplificarea in vitro și detectarea variantei Creutzfeldt - boala Jakob PrPSc , în J. Pathol. , vol. 213, nr. 1, septembrie 2007, pp. 21-6, DOI : 10.1002 / path.2204 , PMID 17614097 .
- ^ Will RG, Ironside JW, Zeidler M, O nouă variantă a bolii Creutzfeldt - Jakob din Marea Britanie , în Lancet , vol. 347, nr. 9006, aprilie 1996, pp. 921–5, DOI : 10.1016 / S0140-6736 (96) 91412-9 , PMID 8598754 .
- ^ a b Pierluigi Gambetti, Boala Creutzfeldt-Jakob (CJD) , pe merckmanuals.com , Manualele Merck: Biblioteca medicală online. Adus 06/04/11 .
- ^ Chakraborty C, Nandi S, Jana S, Boala Prion: o boală mortală pentru plierea greșită a proteinelor , în Current Pharmaceutical Biotechnology , vol. 6, nr. 2, aprilie 2005, pp. 167–77, DOI : 10.2174 / 1389201053642321 , PMID 15853695 .
- ^ a b Obi RK, Nwanebu FC, Prions and Prion Diseases , în African Journal of Clinical and Experimental Microbiology , vol. 9, nr. 1, 2008, pp. 38-52, ISSN 1595-689X . Adus la 20 iunie 2009 .
- ^ Graham A Mackay, Richard SG Knight și James W Ironside,The epidemiology molecular of variant CJD , în International Journal of Molecular Epidemiology and Genetics , 2 (3), nr. 3, 2011, pp. 217-27, PMC 3166149 , PMID 21915360 .
- ^ Manuelidis L, Yu ZX, Barquero N, Banquero N, Mullins B,Cells infected with scrapie and Creutzfeldt-Jakob disease agents produce intracellular 25-nm virus-like particles , in Proceedings of the National Academy of Sciences of the United States of America , vol. 104, n. 6, febbraio 2007, pp. 1965–70, DOI : 10.1073/pnas.0610999104 , PMC 1794316 , PMID 17267596 .
- ^ Mills JL, Schonberger LB, Wysowski DK, Long-term mortality in the United States cohort of pituitary-derived growth hormone recipients , in The Journal of Pediatrics , vol. 144, n. 4, aprile 2004, pp. 430–6, DOI : 10.1016/j.jpeds.2003.12.036 , PMID 15069388 .
- ^ ( EN ) ME Bruce, RG Will, JW Ironside, I. McConnell, D Drummond, A. Suttie, L. McCardle, A. Chree, J. Hope, C. Birkett, S. Cousens, H. Fraser e CJ Bostock, Transmissions to mice indicate that 'new variant' CJD is caused by the BSE agent , in Nature , vol. 389, n. 6650, NY, NY, Nature Publishing Group, 2 ottobre 1997, pp. 498-501, DOI : 10.1038/39057 , ISSN 0028-0836 . URL consultato il 25 aprile 2012 .
- ^ Questions and Answers: Creutzfeldt–Jakob Disease Infection-Control Practices , su Infection Control Practices/CJD (Creutzfeldt–Jakob Disease, Classic) , Centers for Disease Control and Prevention, 4 gennaio 2007. URL consultato il 9 giugno 2007 (archiviato dall' url originale il 17 ottobre 2007) .
- ^ WHO Infection Control Guidelines for Transmissible Spongiform Encephalopathies , su who.int , World Health Organization: Communicable Disease Surveillance and Control, 26 marzo 1999. URL consultato il 9 giugno 2007 .
- ^ McDonnell G, Burke P, The challenge of prion decontamination , in Clinical Infectious Diseases , vol. 36, n. 9, maggio 2003, pp. 1152–4, DOI : 10.1086/374668 , PMID 12715310 .
- ^ Peden AH, Head MW, Ritchie DL, Bell JE, Ironside JW, Preclinical vCJD after blood transfusion in a PRNP codon 129 heterozygous patient , in Lancet , vol. 364, n. 9433, 2004, pp. 527–9, DOI : 10.1016/S0140-6736(04)16811-6 , PMID 15302196 .
- ^ ( EN ) Variant CJD and blood donation ( PDF ), su blood.co.uk , Nayonal blood service - UK. URL consultato il 2 gennaio 2016 (archiviato dall' url originale il 19 aprile 2004) .
- ^ Regan F, Taylor C,Blood transfusion medicine , in BMJ (Clinical Research Ed.) , vol. 325, n. 7356, luglio 2002, pp. 143–7, DOI : 10.1136/bmj.325.7356.143 , PMC 1123672 , PMID 12130612 .
- ^ In-Depth Discussion of Variant Creutzfeld-Jacob Disease and Blood Donation , su redcross.org , American Red Cross. URL consultato il 20 giugno 2009 (archiviato dall' url originale il 30 dicembre 2007) .
- ^ ( DE ) Permanent exclusion criteria , su blutspendehamburg.de , Blutspendedienst Hamburg. URL consultato il 20 giugno 2009 (archiviato dall' url originale il 18 luglio 2011) .
- ^ ( PL ) INFORMACJE DLA KRWIODAWCÓW (Informazioni per i donatori di sangue) , su rckik-warszawa.com.pl , Regionalne Centrum Krwiodawstwa i Krwiolecznictwa w Warszawie. URL consultato il 2 gennaio 2016 (archiviato dall' url originale il 1º settembre 2007) .
- ^ Servizio Trasfusionale CRS della Svizzera Italiana | Chi può donare il sangue? , su svizzera-italiana.trasfusione.ch . URL consultato il 21 settembre 2018 .
- ^ By Rachael Rettner, Blood test may screen for human form of mad cow , MSNBC , 3 febbraio 2011. URL consultato il 9 febbraio 2011 .
- ^ a b Zerr I, Kallenberg K, Summers DM, et al., Updated Clinical Diagnostic Criteria for sporadic Creutzfeldt-Jakob Disease in Brain, vol. 132 (10), 2009, pp. 2659-2668 DOI:10.1093/brain/awp191
- ^ Geoffrey S. Young, G, F, M, H, L, L, W e L, Diffusion-Weighted and Fluid-Attenuated Inversion Recovery Imaging in Creutzfeldt–Jakob Disease: High Sensitivity and Specificity for Diagnosis , in American Journal of Neuroradiology , vol. 26, n. 6, American Society of Neuroradiology, giugno–luglio 2005, pp. 1551–1562, PMID 15956529 . URL consultato il 30 ottobre 2007 .
- ^ Henriette J. Tschampa, M, F, P, S e U, Thalamic Involvement in Sporadic Creutzfeldt–Jakob Disease: A Diffusion-Weighted MR Imaging Study , in American Journal of Neuroradiology , vol. 24, n. 5, American Society of Neuroradiology, 1º maggio 2003, pp. 908–915, PMID =12748093. URL consultato il 30 ottobre 2007 .
- ^ Sanchez-Juan, P., Green, A., Ladogana, A., et al.,CSF tests in the differential diagnosis of Creutzfeldt-Jakob disease , in Neurology , vol. 67, n. 4, 2006, pp. 637–643, DOI : 10.1212/01.wnl.0000230159.67128.00 , PMID 16924018 .
- ^ Tattum, MH, Jones, S., Pal, S., Khalili-Shirazi, A., Collinge, J., Jackson, G., A highly sensitive immunoassay for the detection of prion-infected material in whole human blood without the use of proteinase K , in Transfusion , vol. 50, n. 12, AABB, dicembre 2010, pp. 2619–2627, DOI : 10.1111/j.1537-2995.2010.02731.x , PMID 20561299 .
- ^ Detecting Prions in Blood ( PDF ), in Microbiology Today , agosto 2010, p. 195. URL consultato il 21 agosto 2011 .
- ^ SOFIA: An Assay Platform for Ultrasensitive Detection of PrP Sc in Brain and Blood ( PDF ), su bionosis.com , SUNY Downstate Medical Center. URL consultato il 19 agosto 2011 .
- ^ a b Sternberg's Diagnostic Surgical Pathology, 5th edition.
- ^ Pathology of Degenerative CNS Diseases
- ^ Belay ED, Schonberger LB, Variant Creutzfeldt–Jakob disease and bovine spongiform encephalopathy , in Clin. Lab. Med. , vol. 22, n. 4, 2002, pp. 849–62, v–vi, DOI : 10.1016/S0272-2712(02)00024-0 , PMID 12489284 .
- ^ Teenager with vCJD stable , su news.bbc.co.uk , London, BBC News, 13 dicembre 2004. URL consultato il 1º gennaio 2007 .
- ^ Ian Bone, Lay summary of a report by Professor Ian Bone: Intraventricular Pentosan Polysulphate in Human Prion Diseases - A study of Experience in the United Kingdom ( PDF ), su mrc.ac.uk , Medical Research Council - UK. URL consultato il 19 ottobre 2014 .
- ^ Use of Pentosan Polysulphate in the treatment of, or prevention of, vCJD , su dh.gov.uk , Department of Health:CJD Therapy Advisory Group. URL consultato il 30 ottobre 2007 .
- ^ Rainov NG, Tsuboi Y, Krolak-Salmon P, Vighetto A, Doh-Ura K, Experimental treatments for human transmissible spongiform encephalopathies: is there a role for pentosan polysulfate? , in Expert opinion on biological therapy , vol. 7, n. 5, 2007, pp. 713–26, DOI : 10.1517/14712598.7.5.713 , PMID 17477808 .
- ^ Pfeifer A, Eigenbrod S, Al-Khadra S, Lentivector-mediated RNAi efficiently suppresses prion protein and prolongs survival of scrapie-infected mice , in The Journal of Clinical Investigation , vol. 116, n. 12, dicembre 2006, pp. 3204–10, DOI : 10.1172/JCI29236 , PMC 1679709 , PMID 17143329 .
Voci correlate
- Encefalopatia spongiforme bovina
- Encefalopatia spongiforme trasmissibile
- Kuru (malattia)
- Nuova variante di MCJ
- Prione
Altri progetti
-
 Wikimedia Commons contiene immagini o altri file su malattia di Creutzfeldt-Jakob
Wikimedia Commons contiene immagini o altri file su malattia di Creutzfeldt-Jakob
Collegamenti esterni
- ( EN ) Malattia di Creutzfeldt-Jakob / Malattia di Creutzfeldt-Jakob (altra versione) , su Enciclopedia Britannica , Encyclopædia Britannica, Inc.
- Associazione Italiana Encefalopatie da Prioni - AIEnP onlus , su aienp.it .
| Controllo di autorità | Thesaurus BNCF 54342 · LCCN ( EN ) sh85069294 · GND ( DE ) 4220958-4 · BNF ( FR ) cb12064993z (data) · NDL ( EN , JA ) 00575235 |
|---|