Boala Wilson
| Boala Wilson | |
|---|---|
 | |
| Boala rara | |
| Cod. SSN | RC0150 |
| Specialitate | endocrinologie |
| Etiologie | genetică |
| Clasificare și resurse externe (EN) | |
| ICD-9 -CM | 275.1 |
| OMIM | 277900 |
| Plasă | D006527 |
| MedlinePlus | 000785 |
| eMedicină | 183456 și 1153622 |
| GeneReviews | Prezentare generală |
| Sinonime | |
| degenerescenta hepatolenticulară degenerescenta lenticulara progresiva | |
| Eponime | |
| Samuel Alexander Kinnier Wilson | |
Boala Wilson (denumită anterior și boala Wilson [1] ), sau degenerescența hepatolenticulară , este o boală genetică , transmisă în mod autosomal recesiv , care determină acumularea de cupru în țesuturi ; simptomele se manifestă la nivel neurologic - psihiatric și mai ales la nivelul ficatului . La copii, debutul este adesea cu simptome hepatice, în timp ce la adulți, simptomele neurologice încep mai întâi. [2] Poate apărea între 5 și 40 de ani, iar debutul precoce corespunde unui curs mai grav, periculos și rapid. [3] Simptomele apar de obicei între 6 și 20 de ani, deși simptomele timpurii au fost descrise în unele cazuri la pacienți mult mai în vârstă. [4]
Condiția se datorează unei mutații a proteinei bolii Wilson ( gena ATP7B). O singură copie anormală a genei este prezentă la o sută de persoane, fără a provoca niciun simptom, fiind patologia cauzată de o genă recesivă (aceștia sunt purtători sănătoși ai bolii). Dacă o persoană moștenește gena de la ambii părinți, riscă să dezvolte boala. Boala Wilson are o incidență de 2,66 / 100 000 cu o prevalență de 6,21 / 100 000. [4] Boala poartă numele lui Samuel Alexander Kinnier Wilson (1878-1937), neurolog englez care a descris prima dată afecțiunea în 1912. [5]
Fără tratament, boala poate fi ușor fatală în câțiva ani, dar există medicamente eficiente care o pot controla; tratamentul implică utilizarea unor medicamente chelatoare care reduc absorbția cuprului și elimină excesul de cupru din organism, medicamente de întreținere, uneori fizioterapie și o dietă adecvată săracă în cupru, dar ocazional este necesar și un transplant hepatic în caz de insuficiență hepatică severă . [6] Datorită variabilității mari a simptomelor și a evoluției, diagnosticul este rareori oportun, așa că pacienții așteaptă chiar și ani, cu simptome agravate, înainte de a ști că sunt afectați și pot fi tratați. Cu toate acestea, dacă este tratată în mod adecvat, boala regresează în mare măsură și nu reduce speranța de viață a pacienților, care rămâne identică cu cea a populației sănătoase, precum și menținerea unei bune calități a vieții ; totuși, tratamentul și monitorizarea medicamentelor trebuie să continue în mod constant și cronic pe toată durata vieții. [2]
fundal

Boala poartă numele medicului britanic Samuel Alexander Kinnier Wilson (1878-1937), neurolog care a descris starea, inclusiv modificări patologice la nivelul creierului și ficatului, în 1912 . [5] Munca lui Wilson fusese precedată de rapoartele neurologului german Carl Westphal care, în 1883 , a definit starea ca „pseudoscleroză”, de cele ale neurologului britanic William Gowers (în 1888 ) și de studiile efectuate în 1898 de Adolph Strümpell care a observat ciroză hepatică. [7] În 1958 , neuropatologul John Nathaniel Cumings , a studiat corelația dintre acumularea de cupru atât în ficat, cât și în creier. [8] Prezența hemolizei a fost observată în 1967 . [9]
Cuming împreună și simultan cu neurologul din Noua Zeelandă Derek Denny-Brown , care a lucrat în Statele Unite , a identificat pentru prima dată, în 1951 , un tratament eficient cu Dimercaprol . [10] [11] Medicamentul a fost injectat și a fost una dintre primele terapii disponibile în neurologie, un specialist care, din punct de vedere istoric, a fost capabil să observe și să diagnosticheze, dar care a avut puține tratamente eficiente. [7] [12] Primul medicament eficient ca agent de chelare orală, penicilamina , a fost descoperit în 1956 de neurologul britanic John Walshe . [13] În 1982 , Walshe a introdus și trientina [14] și a fost primul care a dezvoltat tetratiomolibdat pentru uz clinic [15] . Terapia cu acetat de zinc a apărut pentru prima dată în Olanda , unde Schouwink și Hoogenraad au folosit-o în 1961 și respectiv în 1970 , această terapie a fost dezvoltată ulterior de Brewer și colegii de la Universitatea din Michigan . [16] [17]
Baza genetică a bolii Wilson și legătura cu mutațiile genei ATP7B au fost clarificate în anii 1980 și 1990 de diferite grupuri de cercetare; doi pediatri italieni, Enrico Parano și Lorenzo Pavone, au fost printre descoperitorii genei. [18] [19]
semne si simptome
Cuprul se acumulează în principal în ficat și creier ; aceasta este urmată de manifestările hepatice și, respectiv, neuropsihiatrice, care reprezintă principalele semne care conduc la diagnostic . [6] Pacienții cu probleme hepatice apelează, de obicei, la analize medicale mai devreme, în general în copilărie sau adolescență, decât cei cu simptome neurologice sau psihiatrice, care în schimb apelează la aceasta de la vârsta de douăzeci și peste, când tabloul clinic este deja mai compromis. Unele persoane cu boală sunt identificate numai pentru că sunt înrudite cu persoane care au fost deja diagnosticate cu boala Wilson; În urma testului, se constată adesea că mulți dintre acești pacienți au prezentat deja simptome ale bolii fără a fi diagnosticați. [20]
Afectarea ficatului

Ficat tulburări pot prezenta oboseală, sângerări , abdominale dureri sau confuzie ( din cauza encefalopatiei hepatice , vezi simptome neuropsihiatrice ale bolii) și portalul hipertensiune . În plus, în cazul în care presiunea din vena portală crește considerabil, apar varice esofagiene (vasele de sânge care sângerează în esofag ), splenomegalie ( mărirea splinei ) și ascită (acumularea de lichid în cavitatea abdominală ). La examinare, pot fi detectate semne de disconfort hepatic cronic, cum ar fi telangiectasia (vasele de sânge distinse, de obicei pe piept ). Ciroza hepatică , cu fibroză , este adesea diagnosticată la pacienții care raportează simptome. Deși majoritatea persoanelor cu ciroză prezintă un risc mai mare de a dezvolta cancer la ficat , acest risc este relativ scăzut la pacienții cu boala Wilson. [6]
Aproximativ 5% dintre pacienți sunt diagnosticați numai după afecțiuni hepatice bruște, adesea în contextul anemiei hemolitice (anemie datorată distrugerii excesive a globulelor roșii ). Acest lucru duce la producția anormală de proteine și la defecte ale metabolismului hepatic. Acest metabolism deviat duce la acumularea de deșeuri, cum ar fi amoniacul , în fluxul sanguin. Dacă acest produs irită creierul, pacientul dezvoltă encefalopatie hepatică (care poate duce la comă , până la edem cerebral periculos). [6] Anemia provoacă paloare , oboseală , tahicardie , dispnee la efort, icter și colorare anormală a urinei. Rezultatul final este insuficiența hepatică acută sau cronică severă.
Simptome neuropsihiatrice

Aproximativ jumătate dintre pacienții cu boala Wilson au probleme neurologice sau psihiatrice. Majoritatea pacienților prezintă inițial o deteriorare ușoară reversibilă a abilităților cognitive, împreună cu modificări comportamentale. Aceasta este urmată de simptome neurologice specifice, adesea sub formă de parkinsonism [21] : rigiditate crescută, hiperton , în cazuri severe camptocormia , adică postură tipică înainte cu membrele flectate , trăsătură Gowers / festinatio și încetinirea mișcărilor comune, cu sau fără o tipic tremor în mâinile în repaus, salivare , expresiile faciale mascate (amimia); defectele articulației vorbirii ( disartrie ) sunt frecvente; există, de asemenea, ataxie (lipsă de coordonare) și distonie temporară, adică mișcări bruște și necontrolate ale părților corpului: contracții musculare involuntare [22] [23] similare cu clonele și alte diskinezii, cum ar fi posturi bizare, extensii forțate, răsucire în jurul unui articulație simplă, crampe , spasme , contracturi , tremor [24] în mișcare, de obicei la nivelul capului sau al membrelor superioare, hipokinezie , miotonie , sindromul picioarelor neliniștite / acatisie . [25]
Cu toate acestea, cele mai frecvente simptome neurologice în boala Wilson sunt convulsiile și migrenele . [6]
Uneori apar dificultăți de înghițire ( disfagie ), diplopie , oboseală musculară și generală, astenie , dureri musculare , disautonomie , sincopă , dificultate la scris ( disgrafie și reducerea dimensiunii scrisului, așa-numita micrografie parkisoniană ). [26] Poate, de asemenea, să apară rar cu simptome de polineuropatie aparentă. [27]
Problemele psihiatrice datorate bolii Wilson pot include modificări de comportament, cum ar fi depresia , anxietatea , confuzia mentală , până la psihoză ( delir , paranoia , halucinații , putând imita schizofrenia și tulburarea schizoafectivă ) și în stadiul avansat simptome similare cu acele simptome ale subcorticalului demență și demență frontotemporală ( tulburări de dispoziție de origine non-psihiatrică, impulsivitate, judecată afectată, tendință spre promiscuitate și hipersexualitate sau apatie și hiposexualitate , disfuncție executivă cu planificare și luare a deciziilor slabe, gândire lentă, pierdere de memorie , dar fără semne de afazie , apraxia [ 28] sau agnozie [29] prezentă în demențe adevărate). [6] În cele din urmă, poate apărea paralizie pseudobulbară .
Simptomele psihiatrice sunt frecvent observate împreună cu simptome neurologice, rareori manifestându-se singure. Aceste simptome nu sunt adesea bine definite și pot fi atribuite altor cauze. Din acest motiv, diagnosticul bolii Wilson este rar pus atunci când sunt prezente doar simptome psihiatrice, deoarece medicul poate fi indus în eroare în gândirea unei patologii pur psihice. [21]
Alte organe
Mai multe organe sunt implicate în acumularea de cupru în boala Wilson: [30]
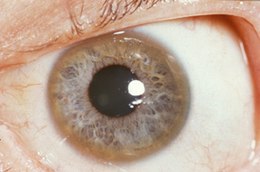
- ochi : inele Kayser-Fleischer . Acestea se datorează depunerii de cupru în membrana Descemet a corneei . Acestea nu apar la toate persoanele și pot fi vizibile la examinarea lămpii cu fantă . Boala Wilson este, de asemenea, asociată cu cataracta , pigmentarea capsulei maro sau verde. Inelele Kayser-Fleischer apar în 66% din cazuri; [20]
- rinichi și sistemul osos : acidoză tubulară renală tip "distală" , boală care duce la nefrocalcinoză ( nefropatie metabolică prin acumulare de calciu în rinichi), slăbirea oaselor (datorită pierderii de calciu și fosfat ) cu osteoporoză , osteomalacie și osteoartrită (prezența artralgii , osteofite , artroza vertebrală a spinării coloanei provocând daune la rădăcinile nervoase) [31] [32] , și ocazional aminoacidurie (pierderea aminoacizilor în urină, necesară pentru sinteza proteinelor ); [6]
- inima : cardiomiopatia dobândită (slăbiciune miopatică a mușchiului inimii) este o problemă rară, dar recunoscută în boala Wilson și poate duce la insuficiență cardiacă și aritmii cardiace (episoade de bătăi neregulate ale inimii); [6]
- hormoni și sistemului endocrin : hipoparatiroidismul (insuficienta a paratiroide glandelor, ceea ce duce la redus plasmatic de calciu niveluri ), infertilitate , amenoree , întârziat pubertate și obișnuită avorturi spontane ; rareori hipotiroidism ; [6] [33]
- Pielea : uneori lunile de culoare unghiilor ceruleană , xeroză (piele uscată), pigmentare anormală, pielea ușor infectată ; [34]
- diverse: anorexie , inapetență, pierderea gustului ( ageusia ), uscăciune și subțierea părului ( alopecie ușoară), agranulocitoză , tulburări gastrointestinale și digestive ca o consecință a problemelor hepatice, tulburări ale mucoasei , ușurința anafilaxiei alergice . [35]
Genetica

Gena responsabilă de boala Wilson ( ATP7B ) a fost identificată și mapată la cromozomul 13 (13q14.3) și este exprimată în principal în ficat, rinichi și placentă . Gena codifică o ATPază de transport de tip P care are funcția de a regla efluxul de cupru din hepatocit în bilă și legarea acestuia de ceruloplasmină . [6] Mutațiile sunt detectate la 90% dintre subiecți. Majoritatea (60%) sunt mutații homozigote ale genei ATP7B (două copii anormale) și 30% au o singură copie anormală (mutație heterozigotă ). Zece la sută nu au nicio mutație detectabilă. [20]
Deși au fost descrise 300 de mutații ale genei ATP7B, în majoritatea cazurilor de boală Wilson din populație, acestea se datorează unui număr mic de mutații specifice pentru acea populație dată. De exemplu, la populațiile occidentale, mutația genei H1069Q (substituirea unei histidine cu o glutamină în poziția 1069 a proteinei) este prezentă în 37-63% din cazuri, în timp ce în China această mutație este foarte rară, în timp ce R778L ( arginina în locul leucinei în poziția 778) se găsește mai frecvent. Se știe relativ puțin despre impactul relativ al diferitelor mutații, deși mutația H1069Q pare, potrivit unor studii, să ducă la apariția problemelor neurologice în viitor. [6] [36]
O modificare a genei normale PRNP poate schimba cursul bolii, întârzierea vârstei de debut și schimbarea simptomelor. De fapt, această genă produce o proteină prionică , care este activă în creier și în alte țesuturi și, de asemenea, pare să fie implicată în transportul cuprului. [37] Rolul genei ApoE a fost inițial suspectat, dar nu a fost confirmat ulterior de studii. [36]
Boala este moștenită într- un mod autosomal recesiv . Pentru transmitere, ambii părinți ai unei persoane afectate trebuie să aibă o genă modificată. Majoritatea nu au antecedente familiale ale bolii. [36] Persoanele cu o singură genă anormală sunt numite purtători ( heterozigoti ) și pot avea anomalii ușoare, nesemnificative din punct de vedere clinic, în metabolismul cuprului. [38]
Boala Wilson este cea mai frecventă cauză a unui grup de boli moștenite care cauzează supraîncărcarea cuprului în ficat. Toate acestea pot provoca ciroza la o vârstă fragedă. Celelalte boli din grup sunt: ciroza infantilă indiană , ciroza infantilă tiroleză endemică și toxicoza idiopatică a cuprului . Cu toate acestea, aceste boli nu sunt legate de mutațiile genei ATP7B. De fapt, de exemplu, ciroza indiană a copiilor a fost legată de mutații ale genei KRT8 și KRT18. [36]
Fiziopatologie
Cuprul este necesar organismului pentru un anumit număr de funcții fiziologice, este mai presus de toate un cofactor pentru funcționarea corectă a unei serii de enzime precum: citocrom-c oxidază , ceruloplasmină , dopamină β-hidroxilază , superoxid dismutază și monofenol monooxigenază . [36]
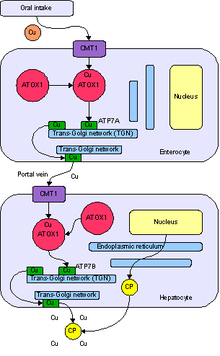
Cuprul pătrunde în organism prin tractul digestiv . O proteină purtătoare:
- CMT1, plasat pe celulele intestinului subțire , transportă cuprul în interiorul lor, unde este legat, parțial cu o anumită metalotioneină și parțial transportat de ATOX1 către aparatul Golgi al enterocitului. Aici, ca răspuns la creșterea concentrațiilor de cupru, o enzimă numită ATP7A eliberează cupru în vena portă și apoi în ficat .
- celulele hepatice posedă și proteina CMT1 care transportă cuprul în interiorul lor, unde este legată de o proteină cu activitate enzimatică feroxidază , ceruloplasmina . Aici, enzima ATP7B o eliberează în sânge sau elimină excesul din ficat, secretându-l în bilă .
Ambele funcții ale ATP7B sunt afectate de boala Wilson. Cuprul se acumulează astfel în țesutul hepatic, ceruloplasmina este încă secretată, dar într-o formă cu deficit de cupru care se degradează rapid în fluxul sanguin. [36]
Atunci când cantitatea de cupru din ficat depășește proteinele care îl leagă în mod normal, daunele oxidative apar printr-un proces cunoscut sub numele de „ reacția Fenton ”. Această oxidare patologică implică apariția hepatitei neinfecțioase, a fibrozei (depunerea țesutului conjunctiv ) și a cirozei. Ficatul eliberează, de asemenea, cupru, care nu este legat de ceruloplasmină, în sânge. Acest cupru liber se răspândește în tot corpul, dar afectează în principal ochii, rinichii și creierul.
În creier, cea mai mare parte a cuprului se depune în ganglionii bazali , în special în putamen și globul pal (denumit în mod colectiv nucleul lenticular ). Se știe că aceste zone participă în mod normal la coordonarea mișcării și joacă un rol semnificativ în procesele neurocognitive, cum ar fi procesarea stimulilor și reglarea dispoziției. Deteriorarea acestor zone (împreună cu o posibilă encefalopatie hepatică) produce în consecință simptomele neuropsihiatrice observate în boala Wilson. [36]
Nu este clar de ce boala Wilson provoacă hemoliză , dar mai multe indicii sugerează că nivelurile ridicate de cupru nelegate de ceruloplasmină au: un efect direct asupra oxidării hemoglobinei , inhibarea furnizorilor de energie enzimatică a celulelor roșii din sânge sau duce la o afectare directă la membrana lor celulară . [39]
Diagnostic

Boala Wilson poate fi suspectată pe baza oricăruia dintre simptomele de mai sus sau atunci când s-a constatat că o rudă apropiată o are. Majoritatea pacienților au teste anormale ale funcției hepatice, cum ar fi creșterea transaminazelor și a bilirubinei . Dacă afectarea ficatului este semnificativă, nivelurile de albumină pot fi scăzute din cauza incapacității celulelor hepatice deteriorate de a produce această proteină. În mod similar, timpul de protrombină (un test de coagulare a sângelui ) poate fi prelungit, deoarece ficatul este incapabil să facă proteinele cunoscute ca factori de coagulare. [6] Nivelurile de fosfatază alcalină sunt relativ scăzute la persoanele bolnave și acest lucru este legat de insuficiența hepatică acută. [40]
Nu există un test complet fiabil pentru diagnosticul bolii Wilson, dar ceruloplasminul plasmatic și nivelurile de cupru, precum și cantitatea de cupru excretată în urină pe o perioadă de 24 de ore, sunt utilizate pentru a afla cantitatea de cupru din organism. . Cu toate acestea, testul standard ideal pentru diagnostic este biopsia hepatică. [6]
Ceruloplasmin
Nivelurile de ceruloplasmină sunt anormal de scăzute (<0,2 g / L) în 80-95% din cazurile de boală. [6] Cu toate acestea, nivelurile normale pot fi prezente la persoanele cu inflamație continuă, deoarece este o proteină de fază acută . Valori scăzute ale ceruloplasminului se găsesc și în boala Menkes și aceruloplasminemia , boli mult mai rare decât boala Wilson. [6] [38]
Ferritin
Ferritina pare a fi ridicată. [41]
Kayser-Fleischer sună
Combinația de simptome neurologice, nivelul scăzut de ceruloplasmină și prezența inelelor Kayser-Fleischer în ochi sunt considerate suficiente pentru diagnosticarea bolii Wilson. În multe cazuri, totuși, sunt necesare investigații suplimentare. [38]
Cupru în urină și ser
Valorile serice ale cuprului sunt scăzute, dar paradoxal sunt ridicate în urină . Pentru examinare, urina este colectată timp de 24 de ore într-o sticlă cu un strat fără cupru. Nivelurile peste 100 µg / 24h (1,6 µmol / 24h) confirmă boala Wilson și nivelurile peste 40 µg / 24h (0,6 µmol / 24h) sunt puternic indicative. [6] Cu toate acestea, nivelurile ridicate de cupru în urină nu sunt unice pentru boala Wilson. Uneori, acestea sunt, de fapt, observate în hepatita autoimună și colestaza (orice boală care împiedică fluxul de bilă de la ficat la intestinul subțire ). [38]
La copii se poate folosi testul penicilaminei . Se administrează o doză orală de 500 mg de penicilamină și se colectează urină de 24 de ore. Dacă acestea conțin mai mult de 1 600 mg (25 micromoli) de cupru, acesta devine un indicator fiabil al bolii Wilson. Cu toate acestea, acest test nu a fost validat pentru utilizare la adulți. [38]
Biopsie hepatică
Odată ce testele au indicat boala Wilson, testul ideal de certitudine este îndepărtarea unei cantități mici de țesut hepatic printr-o biopsie hepatică. Țesutul prelevat este evaluat la microscop pentru a identifica gradul de ficat gras și ciroza. Testele histochimice sunt apoi utilizate pentru a cuantifica cantitatea de cupru și pentru a măsura severitatea acumulării. Un nivel de 250 µg de cupru pe gram de țesut hepatic uscat confirmă boala Wilson. Ocazional pot fi găsite niveluri scăzute de cupru, caz în care combinația rezultatelor biopsiei cu toate celelalte teste efectuate ar putea duce în continuare la un diagnostic formal al lui Wilson. [6]
În stadiile incipiente ale bolii, biopsia prezintă de obicei steatoză (depunerea materialului gras), glicogen crescut în nucleu și zone de necroză (moarte celulară). În boala mai avansată, modificările observate sunt foarte asemănătoare cu cele observate în hepatita autoimună, cum ar fi infiltrarea celulelor inflamatorii, necroza fragmentară și fibroza (țesutul cicatricial). În cele din urmă, în boala avansată, ciroza este principala consecință. În cazul insuficienței hepatice acute, degenerarea celulelor hepatice și distrugerea arhitecturii normale a țesutului duce la un context cirotic. Metodele histochimice pentru detectarea cuprului sunt inconsistente și nesigure și, atunci când sunt realizate singure, sunt considerate insuficiente pentru a stabili un diagnostic precis. [38]
RMN și / sau scanare CT
Dacă sunt prezente simptome neurologice, RMN- ul creierului se face de obicei; poate arăta, de asemenea, „fața uriașă de panda” caracteristică bolii. Imagistica prin rezonanță magnetică nucleară (RMN) și / sau tomografia axială computerizată (CT) poate prezenta modificări ale ganglionilor bazali și ale substanței albe subcorticale sau atrofia unor părți ale acestora. [42]
Teste genetice
Analiza mutațiilor genei ATP7B, precum și a altor gene legate de acumularea de cupru în ficat, poate fi efectuată. Odată confirmată o mutație, este utilă examinarea membrilor familiei. [6]
Diagnostic diferentiat
- Boli neurodegenerative (de exemplu, boala Parkinson cu demență a corpului Lewy , parkinsonism de alte origini, alte demențe , coreea Huntington ), boli neuromusculare (de exemplu, MELAS ) și demielinizante (de exemplu, scleroză multiplă )
- Boala Menkes , sindromul Hallervorden-Spatz și aceruloplasminemia
- Coreea Sydenham
- Deficitul de vitamina B12 (de exemplu, în boala celiacă , anemie pernicioasă )
- Ciroza hepatică de altă origine ( alcoolism , ciroză infantilă indiană , ciroză infantilă tiroleză endemică, toxicoză idiopatică a cuprului)
- Tulburări psihiatrice precum schizofrenia , tulburarea schizoafectivă , psihoză ...
- Nefropatii de alte origini
- Sindromul Wilson (o tulburare legată de hipotiroidism ).
Tratament
Dietă
În general, se recomandă o dietă săracă în alimente care conțin cupru, acestea trebuie evitate: ciuperci , nuci , ciocolată , fructe uscate , ficat de animale și fructe de mare . Nu este necesar, cu excepția fazei acute a cirozei, ca pacientul să excludă complet băuturile alcoolice , deoarece nu este o insuficiență hepatică primară, ci datorită intoxicației cronice cu cupru. [6]
Farmacologic
Există mai multe tratamente medicamentoase disponibile pentru boala Wilson. Unele tind să crească eliminarea cuprului din organism, în timp ce altele împiedică absorbția cuprului din dietă.
În general, penicilamina este primul medicament utilizat. Aceasta leagă cuprul ( chelarea ) și îl conduce să fie excretat în urină. Penicilamina nu este lipsită de contraindicații: aproximativ 20%, de fapt, suferă de efecte secundare sau complicații ale tratamentului, cum ar fi lupusul indus de medicamente (cauzează dureri articulare și erupții cutanate). La cei care au și simptome neurologice, aproape jumătate suferă de o agravare paradoxală a simptomelor. Mentre questo fenomeno si osserva anche in altri trattamenti per la malattia di Wilson, di solito questo è considerato come un'indicazione valida alla sospensione penicillaminica per incominciare un trattamento di seconda linea. [6] [38] I pazienti intolleranti alla penicillamina possono incominciare la terapia con cloridrato di trientina (triethylene tetramine dihydrochloride o 2,2,2-tetramine), che possiede anche proprietà chelanti. Alcuni raccomandano la trientina come trattamento di prima linea, ma l'esperienza con penicillamina è maggiore. [38] Un agente con nota attività per il trattamento nella malattia di Wilson ulteriore è il tetratiomolibdate (tetrathiomolybdate); però questo trattamento è ancora considerato sperimentale, [38] anche se alcuni studi hanno mostrato un effetto benefico. [6]
Una volta che tutti i valori sono ritornati alla normalità, lo zinco (di solito sotto forma di una prescrizione di acetato di zinco , chiamato GALZIN ) può essere utilizzato al posto di chelanti per mantenere stabili i livelli di rame nel corpo. Lo zinco stimola la metallotioneina , una proteina presente nelle cellule dell'intestino che lega il rame e impedisce il loro assorbimento e quindi il trasporto nel fegato. La terapia di zinco è continuata tranne quando avviene una ripresa dei sintomi o quando l'escrezione urinaria di rame aumenta. [38]
Nei rari casi in cui nessuno dei trattamenti per via orale risulti efficace e soprattutto nei casi di grave malattia neurologica, il dimercaprolo risulta necessario. Questo farmaco viene iniettato per via intramuscolare, ogni poche settimane, esso presenta una serie di spiacevoli effetti collaterali come il dolore . [16]
Le persone che sono asintomatiche (ad esempio quelli a cui è stata diagnosticata grazie allo screening familiare oa seguito di test con risultati anomali) vengono generalmente trattate pure, poiché l'accumulo di rame può causare danni nel lungo termine. Non è chiaro se queste persone possano essere trattate meglio con la penicillamina o con l'acetato di zinco. [38]
La terapia fisica
La fisioterapia è utile per quei pazienti che presentano la forma neurologica della malattia. Il trattamento con chelanti del rame può richiedere fino a sei mesi per tornare a lavorare e la terapia fisica può aiutare a far fronte all' atassia , alla distonia e ai tremori, così come può impedire lo sviluppo di contratture che possono derivare da distonia. [43]
Trapianto
Il trapianto di fegato è una cura efficace per la malattia di Wilson, ma è utilizzato solo in scenari di particolare gravità a causa dei numerosi rischi e complicazioni associate alla procedura chirurgica. Viene utilizzato principalmente nelle persone con insufficienza epatica fulminante che non rispondono al trattamento medico o in quelli con avanzata malattia epatica cronica. Il trapianto di fegato è evitato nei casi di grave malattia neuropsichiatrica, in cui la sua efficacia non è stata dimostrata. [6] [38]
Note
- ^ Il termine morbo, dal latino Morbus , "malattia che conduce a morte", è stato storicamente utilizzato per indicare le malattie a decorso fatale , soprattutto perché sconosciute e quindi incurabili. Attualmente è un vocabolo in via di abbandono sia per rispetto del malato, sia perché di molte malattie è stata trovata l'origine e la cura.
- ^ a b Malattia di Wilson su Osservatorio malattie rare
- ^ Malattia di Wilson - Fondazione Telethon
- ^ a b WB. Hu, YZ. Han; BC. Xue; N. Cheng; DY. Sun; DQ. Ye; RM. Yang, [Epidemiological study of hepatolenticular degeneration at Hanshan County, Anhui Province]. , in Zhonghua Yi Xue Za Zhi , vol. 91, n. 13, aprile 2011, pp. 894-7, PMID 21600116 .
- ^ a b Kinnier Wilson SA, Progressive lenticular degeneration: a familial nervous disease associated with cirrhosis of the liver ( PDF ), in Brain , vol. 34, n. 1, 1912, pp. 295–507, DOI : 10.1093/brain/34.4.295 .
- ^ a b c d e f g h i j k l m n o p q r s t u v Ala A, Walker AP, Ashkan K, Dooley JS, Schilsky ML, Wilson's disease , in Lancet , vol. 369, n. 9559, 2007, pp. 397–408, DOI : 10.1016/S0140-6736(07)60196-2 , PMID 17276780 .
- ^ a b Robertson WM, Wilson's disease , in Arch. Neurol. , vol. 57, n. 2, febbraio 2000, pp. 276–7, DOI : 10.1001/archneur.57.2.276 , PMID 10681092 .
- ^ Cumings JN, The copper and iron content of brain and liver in the normal and in hepato-lenticular degeneration ( PDF ), in Brain , vol. 71, Dec, 1948, pp. 410–5, DOI : 10.1093/brain/71.4.410 , PMID 18124738 .
- ^ McIntyre N, Clink HM, Levi AJ, Cumings JN, Sherlock S, Hemolytic anemia in Wilson's disease , in N. Engl. J. Med. , vol. 276, n. 8, febbraio 1967, pp. 439–44, DOI : 10.1056/NEJM196702232760804 , PMID 6018274 .
- ^ Cumings JN, The effects of BAL in hepatolenticular degeneration , in Brain , vol. 74, n. 1, marzo 1951, pp. 10–22, DOI : 10.1093/brain/74.1.10 , PMID 14830662 .
- ^ Denny-Brown D, Porter H, The effect of BAL (2,3-dimercaptopropanol) on hepatolenticular degeneration (Wilson's disease) , in N. Engl. J. Med. , vol. 245, n. 24, dicembre 1951, pp. 917–25, DOI : 10.1056/NEJM195112132452401 , PMID 14882450 .
- ^ Vilensky JA, Robertson WM, Gilman S, Denny-Brown, Wilson's disease, and BAL (British antilewisite [2,3-dimercaptopropanol]) , in Neurology , vol. 59, n. 6, settembre 2002, pp. 914–6, PMID 12297577 .
- ^ Walshe JM, Wilson's disease; new oral therapy , in Lancet , vol. 267, n. 6906, gennaio 1956, pp. 25–6, DOI : 10.1016/S0140-6736(56)91859-1 , PMID 13279157 .
- ^ Walshe JM, Treatment of Wilson's disease with trientine (triethylene tetramine) dihydrochloride , in Lancet , vol. 1, n. 8273, marzo 1982, pp. 643–7, DOI : 10.1016/S0140-6736(82)92201-2 , PMID 6121964 .
- ^ Harper PL, Walshe JM, Reversible pancytopenia secondary to treatment with tetrathiomolybdate , in Br. J. Haematol. , vol. 64, n. 4, dicembre 1986, pp. 851–3, DOI : 10.1111/j.1365-2141.1986.tb02250.x , PMID 3801328 .
- ^ a b Walshe JM, Treatment of Wilson's disease: the historical background , in QJM , vol. 89, n. 7, luglio 1996, pp. 553–5, PMID 8759497 .
- ^ Brewer GJ, Recognition, diagnosis, and management of Wilson's disease , in Proc. Soc. Exp. Biol. Med. , vol. 223, n. 1, gennaio 2000, pp. 39–46, DOI : 10.1046/j.1525-1373.2000.22305.x , PMID 10632959 .
- ^ Bull PC, Thomas GR, Rommens JM, Forbes JR, Cox DW, The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene , in Nat. Genet. , vol. 5, n. 4, 1993, pp. 327–37, DOI : 10.1038/ng1293-327 , PMID 8298639 .
- ^ Tanzi RE, Petrukhin K, Chernov I, et al. , The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene , in Nat. Genet. , vol. 5, n. 4, 1993, pp. 344–50, DOI : 10.1038/ng1293-344 , PMID 8298641 .
- ^ a b c Merle U, Schaefer M, Ferenci P, Stremmel W, Clinical presentation, diagnosis and long-term outcome of Wilson's disease: a cohort study , in Gut , vol. 56, n. 1, 2007, pp. 115–20, DOI : 10.1136/gut.2005.087262 , PMC 1856673 , PMID 16709660 .
- ^ a b Lorincz MT, Neurologic Wilson's disease , in Annals of the New York Academy of Sciences , vol. 1184, 2010, pp. 173–87, DOI : 10.1111/j.1749-6632.2009.05109.x , PMID 20146697 .
- ^ Fahn S. The varied clinical expressions of dystonia. Neurol Clinics. 1984;2:541–554.
- ^ ( EN ) Dystonia - Dystonia Europe , in Dystonia Europe . URL consultato il 30 novembre 2016 .
- ^ Roberto Erro, Ignacio Rubio-Agusti e Tabish A. Saifee, Rest and other types of tremor in adult-onset primary dystonia , in Journal of Neurology, Neurosurgery, and Psychiatry , vol. 85, n. 9, 1º settembre 2014, pp. 965–968, DOI : 10.1136/jnnp-2013-305876 . URL consultato il 29 novembre 2016 .
- ^ MC Trindade, T. Bittencourt, G. Lorenzi‐Filho, RC Alves, DC de Andrade, ET Fonoff, E. Bor‐Seng‐Shu, AA Machado, MJ Teixeira, ER Barbosa, GG Tribl, Restless legs syndrome in Wilson's disease: frequency, characteristics, and mimics , Acta neurologica scandinavica
- ^ La Malattia - Associazione Nazionale Malattia di Wilson ONLUS , su malattiadiwilson.org . URL consultato il 27 maggio 2019 (archiviato dall' url originale il 27 maggio 2019) .
- ^ Keun-Hwa Jung, MD; Tae-Beom Ahn, MD; Beom S. Jeon, MD, PhD, Wilson Disease With an Initial Manifestation of Polyneuropathy , Jama Neurology
- ^ Incapacità di compiere movimenti volontari finalizzati a uno scopo o di comprendere l'uso di oggetti abituali, pur essendo integre l'intelligenza e la motilità
- ^ Disturbo della percezione caratterizzato dal mancato riconoscimento di oggetti, persone, suoni, forme, odori già noti, in assenza di disturbi della memoria e in assenza di lesioni dei sistemi sensoriali elementari.
- ^ Gaetano Crepaldi e Aldo Baritussio, Trattato di medicina interna , PICCIN, 2002, pp. 4687–, ISBN 978-88-299-1642-9 . URL consultato il 27 agosto 2011 .
- ^ MANAGEMENT OF PATIENT WITH HEPATOLIENAL SYNDROME - MANAGEMENT OF PATIENT WITH PORTAL HYPERTENSION - MANAGEMENT OF PATIENT WITH ASCITES
- ^ Wilson Disease - Rare Disease
- ^ Jonathan D. Gitlina, Wilson disease - Clinical presentation, Journal of Gastroenterology
- ^ Seyhan M, Erdem T, Selimoğlu MA, Ertekin V., Dermatological signs in Wilson's disease . Pediatric International Official Journal of the Japan Pediatric Society, 2009 June
- ^ Definitions - Wilson's Disease Association
- ^ a b c d e f g de Bie P, Muller P, Wijmenga C, Klomp LW, Molecular pathogenesis of Wilson and Menkes disease: correlation of mutations with molecular defects and disease phenotypes , in J. Med. Genet. , vol. 44, n. 11, novembre 2007, pp. 673–88, DOI : 10.1136/jmg.2007.052746 , PMC 2752173 , PMID 17717039 .
- ^ Grubenbecher S, Stüve O, Hefter H, Korth C,Prion protein gene codon 129 modulates clinical course of neurological Wilson disease , in Neuroreport , vol. 17, n. 5, 2006, pp. 549–52, DOI : 10.1097/01.wnr.0000209006.48105.90 , PMID 16543824 .
- ^ a b c d e f g h i j k l Roberts EA, Schilsky ML, A practice guideline on Wilson disease ( PDF ) [ collegamento interrotto ] , in Hepatology , vol. 37, n. 6, 2003, pp. 1475–92, DOI : 10.1053/jhep.2003.50252 , PMID 12774027 .
- ^ GR Lee, Chapter 48: acquired hemolytic anaemias resulting from direct effects of infectious, chemical or physical agents , in Lee GR, Foerster J, Lukens J et al. (a cura di), Wintrobe's clinical hematology , vol 1, 10th, Williams & Wilkins, 1999, p. 1298, ISBN 0-683-18242-0 .
- ^ Shaver WA, Bhatt H, Combes B, Low serum alkaline phosphatase activity in Wilson's disease , in Hepatology , vol. 6, n. 5, 1986, pp. 859–63, DOI : 10.1002/hep.1840060509 , PMID 3758940 .
- ^ Hisao Hayashi, Motoyoshi Yano, Yoshikazu Fujita, Shinya Wakusawa, Compound overload of copper and iron in patients with Wilson's disease , in Medical Molecular Morphology , vol. 39, 2006, pp. 121–126, DOI : 10.1007/s00795-006-0326-7 , PMID 16998622 .
- ^ Das SK, Ray K, Wilson's disease: an update , in Nat Clin Pract Neurol , vol. 2, n. 9, settembre 2006, pp. 482–93, DOI : 10.1038/ncpneuro0291 , PMID 16932613 .
- ^ Brewer GJ, Askari FK, Wilson's disease: clinical management and therapy , in Journal of Hepatology , vol. 42, Suppl 1, 2005, pp. 13–21, DOI : 10.1016/j.jhep.2004.11.013 , PMID 15777568 .
Bibliografia
- Joseph C. Segen,Concise Dictionary of Modern Medicine , New York, McGraw-Hill, 2006, ISBN 978-88-386-3917-3 .
- Stephen L. Hauser, Harrison: Neurologia clinica , Casarile (Milano), McGraw-Hill, 2007, ISBN 978-88-386-3923-4 .
- RE. Tanzi, K. Petrukhin; I. Chernov; JL. Pellequer; W. Wasco; B. Ross; DM. Romano; E. Parano; L. Pavone; LM. Brzustowicz, The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene. , in Nat Genet , vol. 5, n. 4, dicembre 1993, pp. 344-50, DOI : 10.1038/ng1293-344 , PMID 8298641 .
- ( EN ) George J. Brewer, Wilson's disease: a clinician's guide to recognition, diagnosis, and management , Springer, 1º maggio 2001, ISBN 978-0-7923-7354-4 .
- ( EN ) Frederick J. Suchy, Ronald J. Sokol e William F. Balistreri, Liver disease in children , Cambridge University Press, 2007, pp. 639–, ISBN 978-0-521-85657-7 .
- ( EN ) John McDonald, Andrew Burroughs e Brian Feagan, Evidence-Based Gastroenterology and Hepatology , John Wiley and Sons, 28 settembre 2010, pp. 493–, ISBN 978-1-4051-8193-8 .
- ( EN ) Anthony S. Fauci, Eugene Braunwald, Dennis Kasper, Stephen Hauser, Dan L. Longo, Harrison's Manual of Medicine , McGraw Hill Professional, 19 marzo 2009, pp. 975–, ISBN 978-0-07-147743-7 .
- ( EN ) Alex J. Mitchell, Neuropsychiatry and behavioural neurology explained , Elsevier Health Sciences, 2004, pp. 171–, ISBN 978-0-7020-2688-1 . URL consultato il 12 giugno 2011 .
- ( EN ) Eugene R. Schiff, Michael F. Sorrell e Willis C. Maddrey, Schiff's diseases of the liver , Lippincott Williams & Wilkins, 2007, pp. 1032–, ISBN 978-0-7817-6040-9 .
- ( EN ) Frances Talaska Fischbach e Marshall Barnett Dunning, A manual of laboratory and diagnostic tests , Lippincott Williams & Wilkins, 2009, pp. 640–, ISBN 978-0-7817-7194-8 .
Linee guida
- ( EN ) LM. Baddour, AE. Epstein; CC. Erickson; BP. Knight; ME. Levison; PB. Lockhart; FA. Masoudi; EJ. Okum; WR. Wilson; LB. Beerman; AF. Bolger, Update on cardiovascular implantable electronic device infections and their management: a scientific statement from the American Heart Association. , in Circulation , vol. 121, n. 3, gennaio 2010, pp. 458-77, DOI : 10.1161/CIRCULATIONAHA.109.192665 , PMID 20048212 .
- ( EN ) NA. Khan, B. Hemmelgarn; RJ. Herman; CM. Bell; JL. Mahon; LA. Leiter; SW. Rabkin; MD. Hill; R. Padwal; RM. Touyz; P. Larochelle, The 2009 Canadian Hypertension Education Program recommendations for the management of hypertension: Part 2--therapy. , in Can J Cardiol , vol. 25, n. 5, maggio 2009, pp. 287-98, PMID 19417859 .
- ( EN ) GL. Myers, RH. Christenson; M. Cushman; CM. Ballantyne; GR. Cooper; CM. Pfeiffer; SM. Grundy; DR. Labarthe; D. Levy; N. Rifai; PW. Wilson, National Academy of Clinical Biochemistry Laboratory Medicine Practice guidelines: emerging biomarkers for primary prevention of cardiovascular disease. , in Clin Chem , vol. 55, n. 2, febbraio 2009, pp. 378-84, DOI : 10.1373/clinchem.2008.115899 , PMID 19106185 .
- ( EN ) DA. Fitzgerald, RJ. Massie; GM. Nixon; A. Jaffe; A. Wilson; LI. Landau; J. Twiss; G. Smith; C. Wainwright; M. Harris, Infants with chronic neonatal lung disease: recommendations for the use of home oxygen therapy. , in Med J Aust , vol. 189, n. 10, novembre 2008, pp. 578-82, PMID 19012558 .
- ( EN ) N. Beydon, SD. Davis; E. Lombardi; JL. Allen; HG. Arets; P. Aurora; H. Bisgaard; GM. Davis; FM. Ducharme; H. Eigen; M. Gappa, An official American Thoracic Society/European Respiratory Society statement: pulmonary function testing in preschool children. , in Am J Respir Crit Care Med , vol. 175, n. 12, giugno 2007, pp. 1304-45, DOI : 10.1164/rccm.200605-642ST , PMID 17545458 .
- ( EN ) W. Wilson, KA. Taubert; M. Gewitz; PB. Lockhart; LM. Baddour; M. Levison; A. Bolger; CH. Cabell; M. Takahashi; RS. Baltimore; JW. Newburger, Prevention of infective endocarditis: guidelines from the American Heart Association: a guideline from the American Heart Association Rheumatic Fever, Endocarditis and Kawasaki Disease Committee, Council on Cardiovascular Disease in the Young, and the Council on Clinical Cardiology, Council on Cardiovascular Surgery and Anesthesia, and the Quality of Care and Outcomes Research Interdisciplinary Working Group. , in J Am Dent Assoc , vol. 138, n. 6, giugno 2007, pp. 739-45, 747-60, PMID 17545263 .
Voci correlate
Altri progetti
-
 Wikiversità contiene risorse su malattia di Wilson
Wikiversità contiene risorse su malattia di Wilson -
 Wikimedia Commons contiene immagini o altri file su malattia di Wilson
Wikimedia Commons contiene immagini o altri file su malattia di Wilson
Collegamenti esterni
- ( EN ) Malattia di Wilson , su Enciclopedia Britannica , Encyclopædia Britannica, Inc.
- http://spazioinwind.libero.it/claudioitaliano/wilson.htm
- http://www.aspe.vb.it/it/morbo_di_wilson.htm
- http://www.sanihelp.it/enciclopedia/scheda/7616.html

| Controllo di autorità | Thesaurus BNCF 50571 · LCCN ( EN ) sh85060301 · GND ( DE ) 4189925-8 · BNF ( FR ) cb124022187 (data) · NDL ( EN , JA ) 00576079 |
|---|